
Abstract
Background
As a life-threatening respiratory syndrome, acute lung injury (ALI) is characterized by uncontrollable inflammatory activities. Semaglutide (SEM) has been identified as an effective anti-inflammatory drug in a variety of diseases. This study intended to explore the functional effect and potential mechanisms of SEM in ALI.
Methods
Lipopolysaccharide (LPS) was used to construct an in vivo ALI model based on Sprague-Dawley (SD) rats and an in vitro ALI model based on human pulmonary artery endothelial cells (HPAECs). Hematoxylin & eosin (H&E) staining and ELISA were applied to evaluate the histopathological changes in pulmonary tissues and detect TNF-α and IL-6 levels. RT-qPCR and Western blotting were used to measure gene and protein expressions in pulmonary tissues and cells. HPAEC viability and apoptosis were evaluated by CCK-8 method and flow cytometry methods.
Results
Semaglutide pretreatment significantly mitigated pulmonary injury, reduced TNF-α and IL-6 production, and led to a decrease in cleaved caspase-3 level and an increase in Bcl-2 level, suggesting SEM could ameliorate LPS-induced ALI in rats. In vitro, SEM increased the proliferative capability and mitigated inflammation and apoptosis in LPS-stimulated HPAECs. In addition, SEM inhibited HDAC5-mediated NF-κB signaling pathway in HPAECs. HDAC5 overexpression or NF-κB signaling activation could partly impair SEM-mediated protective effects against LPS-induced damage to HPAECs.
Conclusion
Semaglutide restrains LPS-induced ALI by inhibiting HDAC5/NF-κB signaling pathway.
Introduction
As a common and severe pulmonary complication with high morbidity and mortality rates, acute lung injury (ALI) poses threat to people’s lives worldwide. 1 ALI is a respiratory syndrome that is often considered an incipient symptom of multiple organ failure and may be caused by multiple disorders, including pneumonia, trauma, and sepsis. 2 Clinically, ALI is characterized by acute inflammation, interstitial edema, diffuse injury in alveolar epithelium, as well as neutrophil accumulation. 3 During ALI onset, the damaged pulmonary endothelial and epithelial cells caused neutrophilic inflammatory responses and increased release of pro-inflammatory cytokines. 4 Despite decades of research on the pathogenetic factors of ALI, the development process of ALI is still ambiguous. 5 Therefore, there is no effective cure for ALI treatment to this day.
It is generally recognized that gram-negative bacterial infection is a major cause of ALI. 6 Lipopolysaccharide (LPS), a vital component of the outer membrane of most gram-negative bacteria, has been identified as an essential activator of inflammatory responses. 7 Therefore, LPS is widely recognized as an important component to construct in vitro and in vivo experimental ALI models for the research and development of novel therapeutic strategies and drugs for ALI. 8
Semaglutide (SEM), an analog of human glucagon-like peptide-1 (GLP-1), is a novel once-weekly drug for diabetes . 9 It has been proven that SEM is not only efficient for diabetes treatment, but also beneficial for the cardiovascular safety of diabetics. 10 SEM is implicated in the regulation of multiple cellular activities, including cell disarray, proliferation, apoptosis, and inflammation. 11 Its antiapoptotic role has been demonstrated in a number of diseases, such as Parkinson’s disease (PD), 12 Alzheimer’s disease (AD), 13 and ischemic stroke. 14 Besides, SEM exhibits anti-inflammatory and protective effects on LPS-induced H9c2 cells. 15 Furthermore, SEM also exerts protective effects against Bleomycin-induced idiopathic pulmonary fibrosis, 16 implying its role in reviving pulmonary function. Therefore, it was suggested that SEM could play a therapeutic role in LPS-induced ALI.
Herein, we mainly investigated the therapeutic effect and molecular mechanism of SEM in LPS-induced ALI. First of all, an in vitro ALI model based on Sprague-Dawley (SD) rats and an in vivo ALI model based on human pulmonary artery endothelial cells (HPAECs) were established with LPS. It was found that SEM could play an ameliorative role in LPS-induced ALI in rats. Moreover, it was also disclosed that SEM could relieve LPS-induced damage to HPAECs via inactivating the HDAC5-mediated NF-κB signaling pathway. Hence, our findings provided novel insights into further research on ALI treatment.
Materials and Methods
Establishment of Lipopolysaccharide-induced acute lung injury model in rats
24 adult male SD rats (8–10 weeks old; 180–220 g) acquired from Laboratory Animal Center of Xuzhou Medical University (Jiangsu, China) were raised in a controlled environment (12 h light/dark cycle; 50%–60% humidity; 25°C), with regular food and water supply.
As Dexmedetomidine (DEX) treatment can ameliorate ALI, 17 DEX treatment is used as positive control. All these SD rats were divided into four groups, namely, Sham (n = 6), ALI (n = 6), ALI+SEM (n = 6), and ALI+DEX (n = 6). Rats in Control group received sterile saline injection subcutaneously. To simulate LPS-induced ALI, rats received LPS injection intratracheally (5 mg/kg). As for ALI+SEM group, rats received SEM injection subcutaneously (25 nM/kg) 30 min ahead of LPS injection. 18 As for ALI+DEX group, rats received DEX (50 μg/kg) injection subcutaneously 30 min ahead of LPS injection. All experimental rats were sacrificed 24h as of LPS injection. Lungs were harvested to collect tissue and bronchoalveolar lavage fluid (BALF) samples. 19
Hematoxylin & eosin staining
The collected lung tissues were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. Then, paraffin-embedded specimens were cut into 5 μm sections for H&E staining. Finally, the sections were examined under a light microscope (Olympus, Japan).
Blood gas level determination
After 24 h as of ALI establishment, rats received endotracheal intubation and were mechanically ventilated with pure oxygen following anesthesia. After ventilation for 20 min, a blood gas analyzer (Radiometer Medical, Denmark) was used to measure the ratio of partial pressure of oxygen (PaO2) to fraction of inspired oxygen (FiO2) (PaO2/FiO2) in arterial blood obtained from the carotid artery.
ELISA assay
TNF-α and IL-6 levels in BALF and HPAEC supernatants were detected via a commercial ELISA kit (R&D Systems) as per standard instructions.
Cell culture, transfection and treatment
HPAECs acquired from American Type Culture Collection (ATCC) were cultured in endothelial cell medium (EGM) in a humidified incubator (5% CO2; 37°C).
HDAC5-pcDNA3.1 vector (oe-HDAC5) and pcDNA3.1 empty vector (Vector) were transfected into HPAECs using Lipofectamine 2000 (Invitrogen) and cultured for 48h before following experiments.
For LPS treatment, HPAECs were treated with LPS (1 μg/mL) for 18 h. For SEM treatment, HPAECs were pretreated with SEM (900 nM) for 30 min before LPS treatment.
CCK-8 assay
CCK-8 assay was performed for HPAEC proliferation detection. Briefly, HPAECs were seeded onto a 96-well plate (5×103 cells/well) and cultivated in a humidified incubator (5% CO2; 37°C) for indicated time. Then, CCK-8 reagent was added (10 μL/well). After incubation for another 2h at room temperature, the optical absorbance was measured with a microplate microscope at 450 nm.
Flow cytometry assay
HPAEC apoptosis was detected with an Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen; CA). Briefly, treated cells were harvested and cultivated with Annexin V and PI at 37°C for 15 min in the dark. A flow cytometer (FACSCalibur, USA) was applied to determine cell apoptosis.
RT-qPCR
Total RNAs of lung tissues and HPAECs were extracted with TRIzol (Invitrogen, USA) and used to generate cDNAs with a reverse transcription kit. Then, qPCR was performed on 7000 Sequence Detection System (Applied Biosystems, USA) with SYBR Green PCR Kit (TransGen, China). GAPDH or U6 was regarded as the internal reference. The fold-change gene expressions were calculated by 2−ΔΔCt method.
Western blotting
Total proteins were extracted from lung tissues and cells via Protein Extraction Reagent Kit (Pierce, USA). Next, Equal amounts of protein lysates were transferred onto PVDF membranes after separation by 12% SDS-PAGE gel. Then, the membranes were blocked with 5% skim milk and cultivated with primary antibodies against (cleaved caspase-3, Bcl-2, HDAC5, P-P65, P65, IκBα, P-IκBα) overnight at 4°C and secondary antibody for 2 h at room temperature. The bands were detected with ECL Plus Western blotting substrate (Thermo Fisher). Finally, the density of the specific protein band was normalized to GAPDH for quantification.
Statistical analysis
Experimental data were presented as mean ± SD and analyzed with GraphPad Prism 7.0 (GraphPad, USA) by Students’ t-test or one-way ANOVA. p <0.05 was deemed significant in statistics.
Results
Semaglutide attenuates Lipopolysaccharide-mediated acute lung injury in rats
To study the protective effect of SEM on LPS-induced ALI in rats, the rats were treated with SEM or DEX 30 min in advance of LPS treatment. The histopathological changes in pulmonary tissues were analyzed by H&E staining. As shown in Figure 1(a), ALI rats manifested severe edema and interstitial infiltration in pulmonary tissues, while the severity of pulmonary injury in ALI rats was dramatically decreased by SEM or DEX pretreatment. In addition, SEM or DEX treatment significantly increased the PaO2/FiO2 ratio in LPS-challenged rats (Figure 1(b)). Pro-inflammatory cytokine (TNF-α and IL-6) levels in BALF samples from these groups were detected by ELISA assay. The results showed that SEM or DEX treatment mitigated inflammation in ALI rats (Figure 1(c) and (d)). Moreover, the levels of pro-apoptotic protein (cleaved caspase-3) and the anti-apoptotic protein (Bcl-2) were detected by Western blotting. In pulmonary tissues from ALI rats, cleaved caspase-3 level was increased while Bcl-2 level was significantly downregulated; however, SEM or DEX pretreatment remarkably decreased cleaved caspase-3 expression and increased Bcl-2 level in ALI rats (Figure 1(e)), suggesting SEM pretreatment might relieve LPS-induced cell apoptosis in pulmonary tissues from ALI rats. The above results indicated that SEM treatment significantly relieved LPS-induced ALI in rats. Semaglutide attenuates Lipopolysaccharide-mediated acute lung injury in rats. (a) SD rats were divided into four groups: Sham (n = 6), ALI (n = 6), ALI+SEM (n = 6), and ALI+DEX (n = 6). H&E staining for pulmonary tissues from each group. (b) Oxygenation index (PaO2/FIO2) of rats from each group. (c and d) ELISA assay for measurement of TNF-α and IL-6 levels in BALF samples. (e) Western blotting for measurement of cleaved caspase-3 and Bcl-2 levels. *p < 0.05; **p < 0.01; ***p < 0.001.
Semaglutide mitigates inflammation and apoptosis of Lipopolysaccharide-induced HPAECs
To investigate the molecular mechanism involved in the protective effects of SEM on LPS-induced ALI, an in vitro ALI model was established by exposing HPAECs to LPS (1 μg/mL) treatment. HPAECs were divided into three groups: Control, LPS, LPS+SEM. According to ELISA assay results, SEM exerted remarkable inhibitory effect on LPS-induced promotive effect on TNF-α and IL-6 secretion (Figures 2(a) and (b)). CCK-8 results manifested that SEM treatment enhanced HPAEC viability, compared with that in LPS group (Figure 2(c)). As indicated by flow cytometry assay, SEM treatment evidently reduced the apoptotic rate of HPAECs (Figure 2(d) and (e)). Western blotting results showed a similar pattern. SEM pretreatment significantly downregulated cleaved caspase-3 level and upregulated Bcl-2 level in LPS-treated HPAECs (Figure 2(f)). Taken together, SEM relieved LPS-mediated damage to HPAECs by enhancing HPAEC viability and inhibiting inflammatory responses and apoptosis. Semaglutide mitigates inflammation and apoptosis of Lipopolysaccharide-induced HPAECs. (a and b) HPAECs were divided into three groups: Control, LPS, LPS+SEM. ELISA assay for measurement of TNF-α and IL-6 levels in supernatants. (c) CCK-8 for HPAEC viability measurement. (d and e) Flow cytometry for detection of HPAEC apoptosis. (f) Western blotting for measurement of cleaved caspase-3 and Bcl-2 levels. *p < 0.05; **p < 0.01.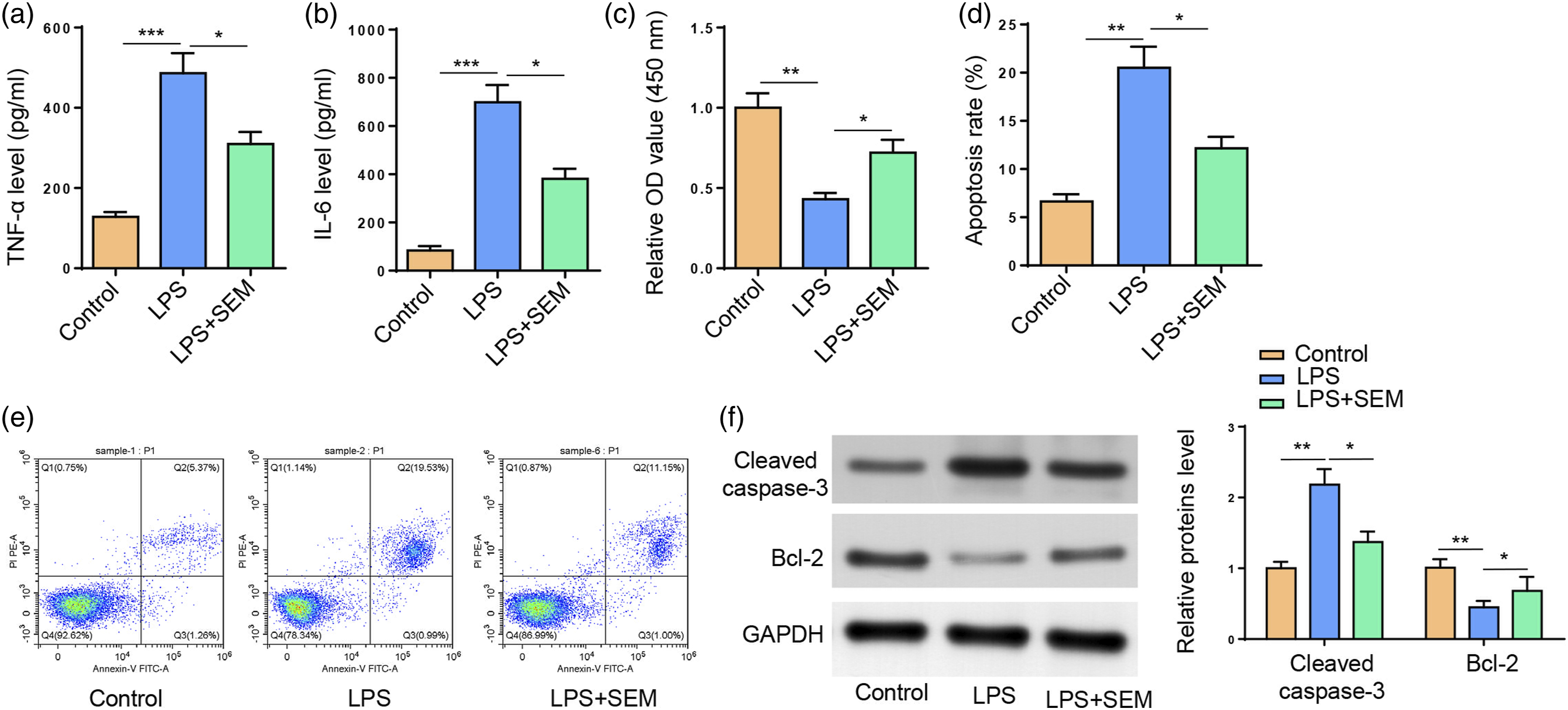
HDAC5 attenuates SEM-mediated protection against Lipopolysaccharide-induced HPAEC damage
It has been demonstrated that HDAC5 actively participates in LPS-induced inflammatory responses.20,21 To examine whether HDAC5 was involved in SEM-mediated protection against LPS-induced ALI, HDAC5 level was detected in pulmonary tissues or HPAECs by IHC or Western blotting. It was revealed that HDAC5 level was upregulated in both pulmonary tissues from ALI rats and LPS-treated HPAECs; whereas SEM treatment significantly downregulated HDAC5 level (Figures 3(a) and (b)). To further investigate the role of HDAC5 in SEM-mediated protective effects on LPS-induced damage to HPAECs, HDAC5 level was overexpressed by transfection with oe-HDAC5 (Figure 3(c)). As indicated in Figure 3(d) and (e), SEM significantly inhibited the TNF-α and IL-6 production in LPS-challenged HPAECs, while HDAC5 upregulation partly abated such a phenomenon. In addition, HDAC5 overexpression dramatically reversed the promoting effects of SEM treatment on the viability of LPS-induced HPAECs (Figure 3(f)). Concurrently, HDAC5 addition also diminished SEM-induced inhibition of the apoptosis in LPS-challenged HPAECs (Figure 3(g) and (h)). In addition, results in Figure 3(i) exhibited that SEM-mediated decrease in cleaved caspase-3 level and increase in Bcl-2 level were neutralized by HDAC5 amplification. Therefore, SEM ameliorated LPS-mediated damage to HPAECs via HDAC5 downregulation. HDAC5 attenuates Semaglutide-mediated protection against Lipopolysaccharide-induced HPAEC damage. (a) IHC for measurement of HDAC5 level in pulmonary tissues. (b) Western blotting for measurement of HDAC5 level in HPAECs. (c) RT-qPCR for measurement of HDAC5 level in HPAECs transfected with Vector or oe-HDAC5. (d and e) HPAECs were divided into four groups: Control, LPS, LPS+SEM, and LPS+SEM+oe-HDAC5. ELISA assay for measurement of TNF-α and IL-6 levels in supernatants. (f) CCK-8 for HPAEC viability measurement. (g and h) Flow cytometry for detection of HPAEC apoptosis. (i) Western blotting for measurement of cleaved caspase-3 and Bcl-2 levels. *p < 0.05; **p < 0.01.
Semaglutide inactivates the NF-κB signaling pathway via downregulating HDAC5 expression in LPS-induced HPAECs
It has been reported that SEM could inhibit the activation of the NF-κB signaling pathway.
22
Also, HDAC5 could activate NF-κB signaling in a murine model of intestinal sepsis.
23
Therefore, it was assumed that SEM might inactivate NF-κB signaling in LPS-induced ALI via regulating HDAC5 expression. In former studies, phorbol myristate acetate (PMA) + Ca2+ ionophore (A23187) treatment has been widely applied for the activation of NF-κB signaling.24-26 Therefore, HPAECs were subject to PMA (25 nmol/L) +A23187 (1 μmol/L) cotreatment to activate NF-κB signaling pathway. HPAECs were divided into Control, LPS, LPS+SEM, LPS+SEM+oe-HDAC5, and LPS+SEM+PMA+A23187 groups. Western blotting was applied to detect HDAC5 and NF-κB-related protein (p-P65, P65, p-IκBα, and IκBα) levels in HPAECs. It was observed that SEM treatment inhibited LPS-induced phosphorylation of P65 and IκBα as well as HDAC5 upregulation in HPAECs, which was remarkably reversed by HDAC5 overexpression or PMA+A23187 cotreatment (Figure 4(a) and (b)). To sum up, SEM could inactivate the NF-κB pathway by downregulating HDAC5. Semaglutide inactivates the NF-κB signaling pathway via downregulating HDAC5 expression in Lipopolysaccharide-induced HPAECs. HPAECs were divided into 5 groups: Control, LPS, LPS+SEM, LPS+SEM+oe-HDAC5, and LPS+SEM+PMA+A23187. (a and b) Western blotting for HDAC5, p-P65, P65, p-IκBα, and IκBα measurement in HPAECs. *p < 0.05; **p < 0.01.
Activation of NF-κB signaling abates SEM-mediated protection against LPS-induced HPAEC damage
To further affirm whether SEM could relieve LPS-treated HPAEC damage by inhibiting NF-κB signaling pathway, HPAECs were divided into Control, LPS, LPS+SEM, and LPS+SEM+PMA+A23187 groups. In contrast to LPS+SEM group, PMA+A23187 cotreatment significantly increased TNF-α and IL-6 secretion in HPAECs (Figures 5(a) and (b)). It was also found that PMA+A23187 could reduce the proliferative capability of HPAECs, in comparison with SEM treatment (Figure 5(c)). Additionally, PMA+A23187 treatment dramatically impaired SEM-induced reduction of apoptosis in LPS-induced HPAECs (Figure 5(d) and (e)), accompanied by elevated cleaved caspase-3 level and decreased Bcl-2 level (Figure 5(f)). The above results substantially evidenced that SEM alleviates LPS-treated inflammation and apoptosis in HPAECs by inactivating the NF-κB signaling pathway. Activation of NF-κB signaling abates Semaglutide-mediated protection against Lipopolysaccharide-induced HPAEC damage. (a and b) HPAECs were divided into four groups: Control, LPS, LPS+SEM, and LPS+SEM+PMA+A23187. ELISA assay for measurement of TNF-α and IL-6 levels in supernatants. (c) CCK-8 for HPAEC viability measurement. (d and e) Flow cytometry for detection of HPAEC apoptosis. (f) Western blotting for measurement of cleaved caspase-3 and Bcl-2 levels. *p < 0.05; **p < 0.01.
Discussion
SEM is a long-acting human GLP-1 analog for diabetes treatment. 27 As discovered by recent studies, SEM also exerts anti-inflammatory and anti-apoptotic effects. 14 For instance, SEM ameliorates testicular dysfunction by attenuating inflammation and reducing apoptosis. 28 Also, SEM plays a neuroprotective role in a murine model of acute ischemic stroke by repressing inflammation and oxidative stress. 29 Moreover, SEM substantially diminishes inflammatory response in an LPS-induced acute inflammation model. 30 In this work, we focused on the protective role of SEM in LPS-treated ALI. Our in vivo experiments showed that SEM pretreatment significantly relieved pulmonary injury in ALI rats, decreased the TNF-α and IL-6 levels in BALF from ALI rats, and caused a decrease in cleaved caspase-3 level and an increase in Bcl-2 level. In vitro, SEM treatment remarkably inhibited the production of TNF-α and IL-6, promoted proliferation, and inhibited apoptosis in LPS-stimulated HPAECs. All these results indicated that SEM could mitigate LPS-induced ALI.
HDAC5 (Histone Deacetylase 5), a member of the histone deacetylase family, could regulate essential cellular processes via histone deacetylation. 21 As demonstrated by accumulating evidence, HDAC5 plays a pro-apoptotic and pro-inflammatory role in human diseases. To cite an instance, HDAC5 knockdown induced neuron apoptosis in an epilepsy model. 31 HDAC5 aggravated inflammation and cell apoptosis in acute pancreatitis. 32 Furthermore, HDAC5 activated the NF-κB pathway via Ghrelin and E2F1 inhibition, thereby exacerbating inflammatory response and apoptosis in LPS-challenged intestinal macrophages. 23 In our work, elevated HDAC5 expression was observed in both in vitro and in vivo ALI models. In addition, SEM treatment significantly reduced HDAC5 level in LPS-treated HPAECs. As shown in functional experiments, SEM treatment enhanced the proliferative ability of LPS-stimulated HPAECs, which was partially abated by HDAC5 overexpression; besides, HDAC5 upregulation reversed the inhibitory effects of SEM treatment on the inflammatory responses and apoptosis of LPS-challenged HPAECs. Therefore, it was affirmed that SEM protected HPAECs against LPS-induced inhibition on cell proliferation and increase in production of pro-inflammatory cytokines and cell apoptosis via downregulating HDAC5 expression.
NF-κB signaling pathway is critical for the regulation of inflammatory response and cellular behaviors including cell proliferation and apoptosis.33,34 It has been demonstrated by previous studies that the inactivation of NF-κB signaling pathway is a strategy for the protection against LPS-induced ALI. For example, Cai et al. 35 found that Astaxanthin inactivated the MAPK/NF-κB pathway to ameliorate LPS-induced ALI . Li et al. 36 disclosed that Alkannin relieved LPS-induced ALI in rats by inhibiting the Rho/ROCK/NF-κB signaling . In addition, Li et al. 37 uncovered that Fraxin exerted protective effects against LPS-induced ALI in rats via inactivation of NLRP3 and NF-κB signaling . Herein, SEM treatment partly neutralized LPS-induced phosphorylation of P65 and IκBα and HDAC5 upregulation; however, HDAC5 overexpression or PMA+A23187 cotreatment dramatically increased the p-P65 and p-IκBα levels and reversed the inhibitory effects on HDAC5 expression, indicating SEM inactivated NF-κB signaling via downregulating HDAC5 expression in HPAECs. Functionally, PMA+A23187 treatment abrogated SEM-induced inhibition of inflammation and apoptosis and promotion of the proliferation of LPS-treated HPAECs. As demonstrated by previous studies, NF-κB stimulation could significantly upregulate the expression of the anti-apoptotic protein, Bcl-2, thereby reducing apoptosis in LPS-induced ALI. 38 Similarly, our data evidenced that PMA+A23187-caused NF-κB activation partly abated SEM-induced cleaved caspase-3 downregulation and Bcl-2 upregulation in LPS-treated HPAECs, which was consistent with the flow cytometry results. All the above results indicated that both HDAC5 and NF-κB signaling were involved in SEM-mediated protection against LPS-induced ALI.
Conclusion
This study, for the first time, identified that SEM mitigated LPS-induced ALI in vitro and in vivo via inactivating HDAC5-dependent NF-κB signaling, providing a theoretical basis for future research and clinical application of SEM in ALI.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.