
Abstract
Long non-coding RNA (lncRNA) small nucleolar RNA host gene 12 (SNHG12) has been reported to participate in the regulation of various nervous system disorders. Bupivacaine (BV), a commonly used local anesthetic, could generate neurotoxicity in neurons. This work intended to investigate the role and specific mechanism of SNHG12 in BV-induced neurotoxicity. In this study, we established an in vitro cell model of BV-induced neurotoxicity by exposing human neuroblastoma cells (SH-SY5Y) to BV. It was found that SNHG12 and NLRX1 levels were gradually downregulated, while miR-497–5p enrichment was upregulated accordingly with the increase of BV concentration. As indicated by functional assays, SNHG12 overexpression promoted cell viability but inhibited cell apoptosis and oxidative stress in BV-treated SH-SY5Y cells. In addition, it was identified that SNHG12 directly targeted miR-497–5p and attenuated BV-induced neurotoxicity via interaction with miR-497–5p. Besides, it was confirmed that SNHG12 could upregulate NLRX1 expression by absorbing miR-497–5p. Moreover, miR-497–5p decreased cell viability and induced cell apoptosis and oxidative stress, which was partly reversed by NLRX1 upregulation. In conclusion, our findings indicated that SNHG12 might relieve BV-associated neurotoxicity by upregulating NLRX1 via miR-497–5p in vitro, providing novel clues and biomarkers for the treatment and prevention of BV-associated neurotoxicity.
Introduction
Bupivacaine (BV) is a kind of anesthetic extensively applied for long-term local anesthesia during labor, surgery, or certain medical procedures. 1 According to previous reports, adverse drug reactions of BV administration could result in severe and even irreversible damage to the central nervous system and cardiovascular system of patients.2–4 Increasing evidence has shown that BV could induce neurotoxicity through diverse signaling pathways.5,6 Nevertheless, few studies have investigated the molecular mechanisms of BV-induced neurotoxicity.
Long non-coding RNAs (lncRNAs) are a group of non-protein-coding RNAs containing more than 200 nucleotides. 7 Many reports have shown that lncRNAs can participate in several biological processes and cellular activities, including apoptosis, 8 proliferation, 9 autophagy, 10 inflammation, 11 and oxidative stress response. 12 Besides, the dysregulation of lncRNAs is associated with the development and progression of various human diseases, including anesthesia-induced neurotoxicity. 13 For example, Yao et al. disclosed that KCNQ1OT1 alleviated anesthesia-induced neurotoxicity through the miR-206/BDNF pathway. 14 Besides, He et al. showed that BACE1-AS knockdown relieved isoflurane-induced neurotoxicity in Alzheimer’s disease via targeting miR-214–3p. 15 The functions of the small nucleolar RNA host gene (SNHG) family in regulating the biological activities of nerve cells have been widely reported. To cite an instance, Zhang et al. demonstrated that SNHG1 aggravated sevoflurane-induced neurotoxicity via interaction with miR-181b. 16 Zhang et al. uncovered that SNHG7 induced inflammatory response and apoptosis in a cell model of Parkinson’s disease (PD). 17 Besides, Pan et al. affirmed the neuroprotective role of SNHG16 in a cellular model of cerebral ischemic/reperfusion (I/R). 18 LncRNA SNHG12, a member of the SNHG family, is located in the p35.3 region of chromosome 1. 19 As revealed by Yan et al. in a former study, SNHG12 could depress neuronal apoptosis caused by oxygen-glucose deprivation (OGD) treatment through upregulating NEGR1 expression via interaction with miR-181a-5p. 20 In addition, Yao et al. found SNHG12 played a neuroprotective role in cerebral I/R injury by inducing autophagy in nerve cells. 21 Also, Yan et al. demonstrated that SNHG12 could regulate neuronal apoptosis and inflammatory responses in PD via the miR-138–5p/NFIB axis. 22 However, the function of SNHG12 in BV-induced neurotoxicity is still unclear.
In the current work, we investigated whether and how SNHG12 participated in BV-induced neurotoxicity. Through a series of functional assays and bioinformatics analysis, we found that SNHG12 played a neuroprotective role against BV-induced neurotoxicity via the miR-497–5p/NLR family member X1 (NLRX1) axis, offering novel targets for preventing or treating BV-induced neurotoxicity.
Materials and Methods
Reagents
The human neuroblastoma cell line (SH-SY5Y) was purchased from Bena Culture Collection (BNCC338056, Suzhou, China). Dulbecco’s modified Eagle medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco Company (Grand Island, USA). Bupivacaine and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St Louis, USA). All reagents used in RT-qPCR were purchased from Invitrogen (USA) or Takara (Japan). All RT-qPCR primers were obtained from Sangon Biotech (Shanghai, China). Other reagents are as follows: CCK-8 reagents (Beyotime, China), Annexin V-fluorescein isothiocyanate (V-FITC) and propidium iodide (PI) (KeyGEN, China), Lactate dehydrogenase (LDH), superoxide dismutase (SOD), malondialdehyde (MDA), and glutathione peroxidase (GSH-Px) commercial kits (Nanjing Jiancheng Bioengineering Institute, China).
Cell culture and BV treatment
SH-SY5Y cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin in an incubator (37°C 5%; CO2). For BV treatment, BV was first dissolved in DMSO. Then, SH-SY5Y cells were incubated with BV at different concentrations (0, 0.5, 1.0, 1.5, or 2.0 mM) for 24 h as described previously. 23 Cells at passage four were used for subsequent analysis.
Cell transfection
SNHG12 overexpression vector (oe-SNHG12), pcDNA3.1 empty vector (Vector), small interfering RNA (siRNA) against SNHG12 (si-SNHG12) or NLRX1 (si-NLRX1), negative control (si-NC), miR-497–5p mimics, miR-497–5p inhibitor, and corresponding negative controls (NC mimics and NC inhibitor) were all synthesized by GenePharma. Then, the above-mentioned plasmids were transfected into SH-SY5Y cells using Lipofectamine 2000 (Invitrogen, USA). After 48 h’ transfection, the cells were harvested for subsequent experiments.
CCK-8 assay
CCK-8 assay was applied for the assessment of cell viability. SH-SY5Y cells were plated into 96-well plates (1 × 104 cells/well) and incubated with different BV concentrations (0, 0.5, 1.0, 1.5, or 2.0 mM) for 24 h. Thereafter, 10 μL CCK-8 solution was added to each well, and the cells were cultured at 37°C for another 2 h. Via a microplate reader (BioTek Instruments, USA), the optical density at 450 nm was detected.
Flow cytometry
Flow cytometry assay was applied to detect cell apoptosis. Briefly, treated SH-SY5Y cells were collected, rinsed with PBS, resuspended in 200 μL Annexin V-binding buffer, and then stained with Annexin V-FITC and PI for 15 min in darkness. The apoptotic cells were measured by an FCM flow cytometer with FlowJo software (BD Bioscience, USA). Q1 = Necrosis, Q2 = Late apoptosis, Q3 = Early apoptosis, Q4 = Viability.
Determination of oxidative stress-associated factors
The SH-SY5Y cell supernatants were harvested after treatment. LDH, SOD, MDA, and GSH-Px levels were measured with corresponding commercial kits (Nanjing Jiancheng Bioengineering Institute, China) as per the standard protocol.
RT-qPCR assay
The total RNA extraction was conducted with TRIzol reagent (Invitrogen, USA). Total RNA was reversely transcribed into cDNA with PrimeScript RT Reagent Kit (Takara, Japan). Thereafter, qPCR was performed with SYBR Green kit on ABI 7500 Real-time PCR system (Applied Biosystems). The relative gene expressions were calculated by 2−ΔΔCt method. GAPDH or U6 was utilized as the internal control. The primers used were listed below: SNHG12: 5′-GGTGCTCCAGGCAATAACT-3′ (forward: F), 5′-CTCCCATACAGTCCGAACAT-3′SNHG12 (reverse: R); NLRX1: 5′-TAGGGCCTTTATCCGTTACCA-3′ (F), 5′-TAAACCACTCGGTGAGGTTCC-3′ (R); BDNF: 5′-GAAAGTCCCGGTATCAAAAG-3′ (F), 5′-CGCCAGCCAATTCTCTTTTTG-3′ (R); GAPDH: 5′-ACCCACTCCTCCACCTTTGAC-3′ (F), 5′-TGTTGCTGTAGCCAAATTCGTT-3′ (R); miR-497–5p: 5′-CCTTCAGCAGCACACTGTGG-3′ (F), 5′-CAGTGCAGGGTCCGAGGTAT-3′ (R); U6: 5′-GCTTCGGCAGCACATATACTAAAAT-3' (F), 5′-CGCTTCACGAATTTGCGTGTCAT-3' (R).
Dual-luciferase reporter assay
DNA fragments of SNHG12 or 3′-UTR NLRX1 with wildtype (WT) or mutant (Mut) binding sequences for miR-497–5p were subcloned into pGL3 vectors to construct corresponding plasmids (SNHG12-WT, SNHG12-MUT, NLRX1-WT, and NLRX1-MUT). The above plasmids were transfected into SH-SY5Y cells, respectively, together with NC mimics or miR-497–5p mimics. After 48 h, the luciferase activity of SH-SY5Y cells in each group was assessed via the Dual-Luciferase Reporter Assay Kit (Promega, USA).
Statistical analysis
Data were acquired from at least three repeated experiments and presented as mean ± standard deviation (SD). GraphPad Prism 6.0 was applied for statistical analysis of differences via one-way ANOVA (more than two groups) or Student’s t-test (two groups). p-value < 0.05 was defined as significant in statistics.
Results
SNHG12 is lowly expressed in an in vitro model of BV-induced neurotoxicity
To explore the regulatory mechanism of BV-induced neurotoxicity, a BV-induced cell model was constructed by treating SH-SY5Y cells with BV at different concentrations (0, 0.5, 1.0, 1.5, or 2.0 mM). As shown by CCK-8 assay, BV treatment markedly suppressed the viability of SH-SY5Y cells (Figure 1(A)). Since 1.0 mM BV concentration could trigger approximately 50% cell growth inhibition in SH-SY5Y cells, 1.0 mM BV concentration was applied for subsequent function assays. Flow cytometry assay exhibited that BV treatment significantly increased apoptosis in SH-SY5Y cells (Figure 1(B)). Next, morphological changes in SH-SY5Y cells were observed. As indicated in Figure 1(C), after exposure to BV (1.0 mM) for 24 h, SH-SY5Y cells became round and were atrophic in shape and incomplete in structure. In addition, the levels of oxidative stress-associated factors (LDH, MDA, SOD, and GSH-Px) were also detected. The results manifested that LDH and MDA levels were elevated, while SOD and GSH-Px levels were declined in BV-treated SH-SY5Y cells (Figure 1(D–G)), indicating that BV treatment led to oxidative stress damage in SH-SY5Y cells. As an important biomarker to evaluate neurotoxicity, the brain-derived neurotrophic factor (BDNF) level was also measured by RT-qPCR. The results showed that the BDNF level in SH-SY5Y cells was dramatically decreased after BV challenge (Figure 1(H)). As evidenced by the attenuated cell viability, the increased cell apoptosis rate and oxidative stress damage, and the decreased BDNF level in SH-SY5Y cells caused by BV treatment, it was shown that BV treatment could lead to neurotoxicity, indicating the successful establishment of the in vitro model of BV-induced neurotoxicity. Furthermore, RT-qPCR results revealed that SNHG12 expression was declined in SH-SY5Y cells with the increase of BV concentration (Figure 1(I)). Therefore, it was speculated that SNHG12 might be involved in the BV-induced cytotoxicity in SH-SY5Y cells.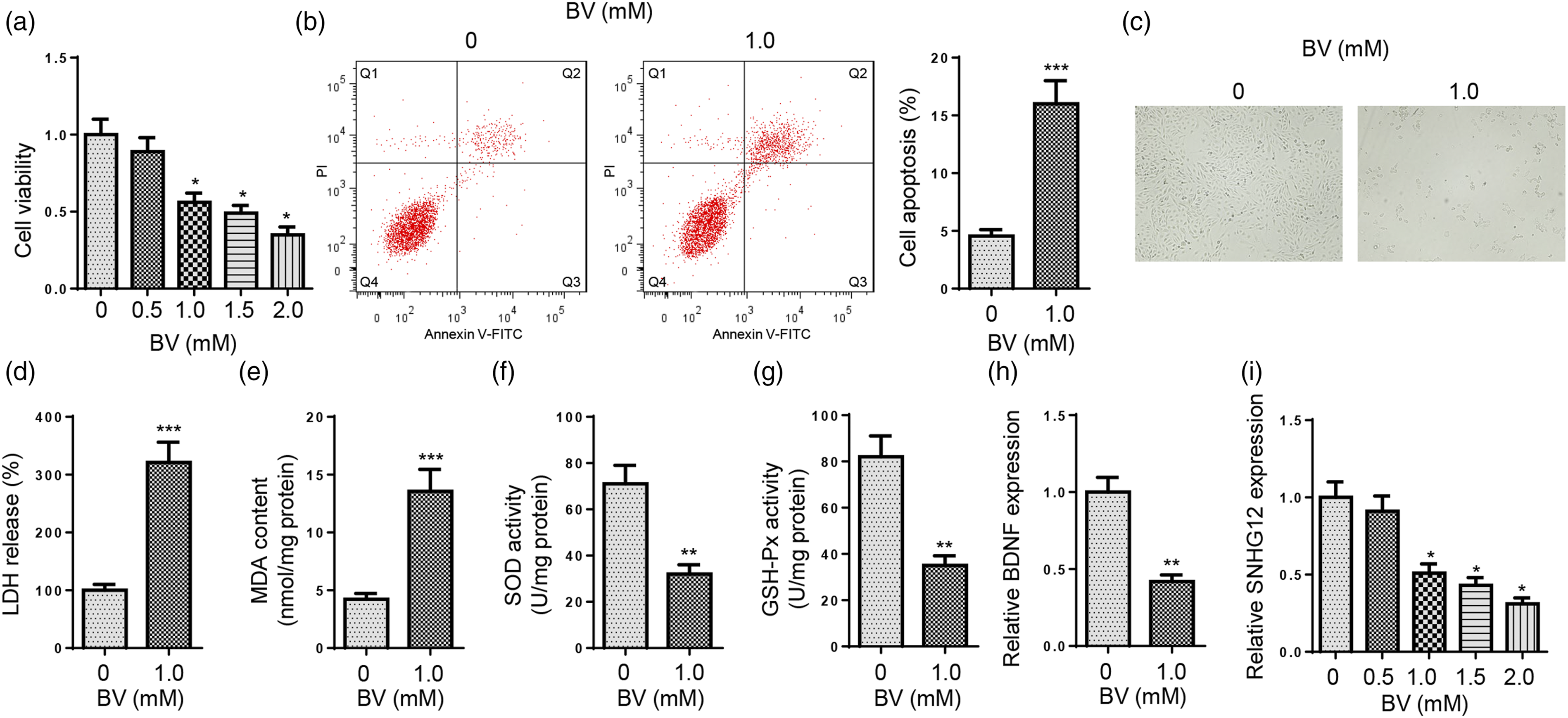
SNHG12 upregulation alleviates the neurotoxicity induced by BV treatment in SH-SY5Y cells
To analyze the regulatory function of SNHG12 in BV-induced neurotoxicity, a series of gain-of-function assays were applied. First of all, SNHG12 was overexpressed in SH-SY5Y cells and the overexpression efficiency was assessed via RT-qPCR (Figure 2(A)). Then, SH-SY5Y cells were firstly subject to BV treatment and then transfected with Vector or oe-SNHG12. As exhibited in Figure 2(B), SNHG12 expression was decreased after BV treatment, while SNHG12 upregulation partly abolished the suppressive effect (Figure 2(B)). CCK-8 assay results showed that BV treatment led to a remarkable inhibitory effect on SH-SY5Y cell viability, while SNHG12 overexpression markedly restored this reduction in SH-SY5Y cells (Figure 2(C)). Flow cytometry showed that the apoptotic rate of SH-SY5Y cells was remarkably enhanced after BV treatment; however, this impact was partly abrogated by SNHG12 upregulation (Figure 2(D)). In addition, SNHG12 addition also distinctly eliminated the impact of BV treatment on LDH, MDA, SOD, and GSH-Px levels (Figure 2(E–H)), suggesting SNHG12 upregulation could eliminate BV-induced oxidative stress damage in SH-SY5Y cells. Moreover, BV-mediated decline in BDNF level was partly reversed by SNHG12 addition (Figure 2(I)). The above data indicated that SNHG12 improved BV-induced neurotoxicity in vitro.
SNHG12 directly targets miR-497–5p
To probe the possible downstream mechanism of SNHG12 in BV-induced neurotoxicity, we applied StarBase 2.0 website to predict the potential target microRNAs (miRNAs) for SNHG12. Under the following conditions (CLIP Data: strict stringency≥5; Pan-Cancer: four cancer types), six downstream microRNAs (miR-199a-5p, miR-320a, miR-424–5p, miR-195–5p, miR-1301–3p, and miR-497–5p) were screened by StarBase website. As indicated by RT-qPCR, only miR-497–5p expression was remarkably declined in SNHG12-overexpressed SH-SY5Y cells (Figure 3(A)). According to a previous study by Chen et al., miR-497–5p could aggravate ketamine-induced neuronal injury via inactivating the TrkB/PI3K/Akt pathway,
24
suggesting miR-497–5p could exacerbate anesthetic-induced neurotoxicity. Therefore, miR-497–5p was chosen as a target of SNHG12 in this study. The binding site between miR-497–5p and SNHG12 was provided by the StarBase website (Figure 3(B)). Next, miR-497–5p overexpression efficiency in SH-SY5Y cells was verified by RT-qPCR (Figure 3(C)). Dual-luciferase reporter assay manifested that the luciferase activity of SNHG12-WT group was remarkably decreased after miR-497–5p upregulation, while the luciferase activity of SNHG12-MUT group showed no significant difference (Figure 3(D)), confirming miR-497–5p as a downstream target of SNHG12 in SH-SY5Y cells. In addition, RT-qPCR results revealed that miR-497–5p level was upregulated with the increase of BV concentration (Figure 3(E)). Next, SNHG12 was knocked down in SH-SY5Y cells, and the knockdown efficiency was confirmed (Figure 3(F)). The results of RT-qPCR assay disclosed that miR-497–5p expression in BV-induced SH-SY5Y cells was reduced after SNHG12 upregulation and increased after SNHG12 downregulation (Figure 3(G)). Taken together, SNHG12 could negatively regulate miR-497–5p expression in SH-SY5Y cells.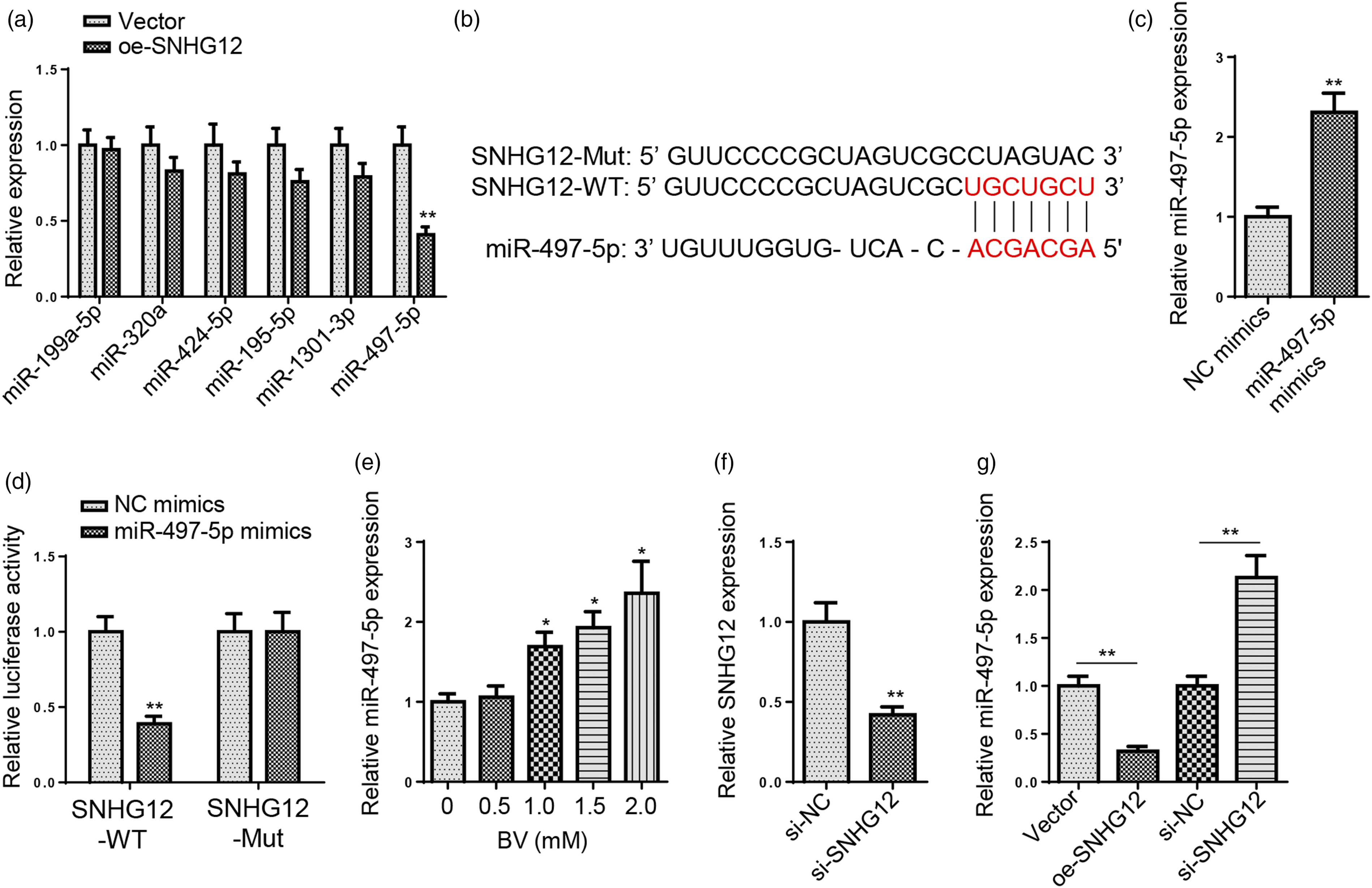
SNHG12 mitigates BV-induced neurotoxicity via miR-497–5p
To study whether SNHG12 regulated BV-induced neurotoxicity via miR-497–5p, SH-SY5Y cells were firstly subject to BV treatment and then transfected with Vector, SNHG12, SNHG12+NC mimics, or SNHG12+miR-497–5p mimics. RT-qPCR results exhibited that miR-497–5p upregulation partially abolished the inhibitory effect of SNHG12 upregulation on miR-497–5p expression (Figure 4(A)). In addition, SNHG12 upregulation promoted the viability of BV-treated SH-SY5Y cells, which was neutralized after miR-497–5p overexpression (Figure 4(B)). Also, miR-497–5p upregulation partly rescued the inhibitory effect of SNHG12 addition on BV-induced SH-SY5Y cell apoptosis (Figure 4(C)). In addition, the suppressive impact of SNHG12 overexpression on BV-induced oxidative damage in SH-SY5Y cells was also neutralized by miR-497–5p amplification, as indicated by the variation of oxidative stress-associated factors (LDH, MDA, SOD, and GSH-Px) (Figure 4(D–G)). Furthermore, the promoting effect of SNHG12 upregulation on BDNF level was reversed by miR-497–5p addition (Figure 4(H)). To sum up, SNHG12 upregulation relieved BV-induced neurotoxicity in SH-SY5Y cells, while miR-497–5p addition partly offset such a mitigative effect, implying SNHG12 regulated BV-induced neurotoxicity in vitro via interaction with miR-497–5p.
NLRX1 is directly targeted by miR-497–5p
To further explore the downstream target of miR-497–5p in BV-induced neurotoxicity, four databases (microT, miRanda, miRmap, and PITA) were employed to predict the target of miR-497–5p. 11 candidate genes (FAM189 B, SINHCAF, ZNF622, SPTBN2, AP2B1, COP1, WIPI2, PISD, SSRP1, NLRX1, and KDSR) were screened (Figure 5(A)). It was demonstrated by Chen et al. that NLR family member X1 (NLRX1) upregulation could relieve trauma-induced brain injury by promoting proliferation and decreasing apoptosis in neurons,
25
indicating its neuroprotective functions. Therefore, NLRX1 was chosen. The binding site between miR-497–5p and NLRX1 was as shown in Figure 5(B). Dual-luciferase reporter assay revealed that the relative luciferase activity of NLRX1-WT was significantly reduced in the miR-497–5p group (Figure 5(C)). As shown in Figure 5(D), NLRX1 expression was substantially reduced with the increase of BV concentration. Then, miR-497–5p inhibition efficiency was verified by RT-qPCR (Figure 5(E)). As revealed by RT-qPCR assay, NLRX1 level in BV-challenged SH-SY5Y cells was markedly reduced after miR-497–5p upregulation and increased after miR-497–5p silencing (Figure 5(F)). Furthermore, NLRX1 expression in BV-treated SH-SY5Y cells was significantly declined after SNHG12 knockdown and dramatically upregulated after SNHG12 addition (Figure 5(G)). In sum, NLRX1 expression in BV-challenged SH-SY5Y cells was negatively regulated by miR-497–5p and positively regulated by SNHG12.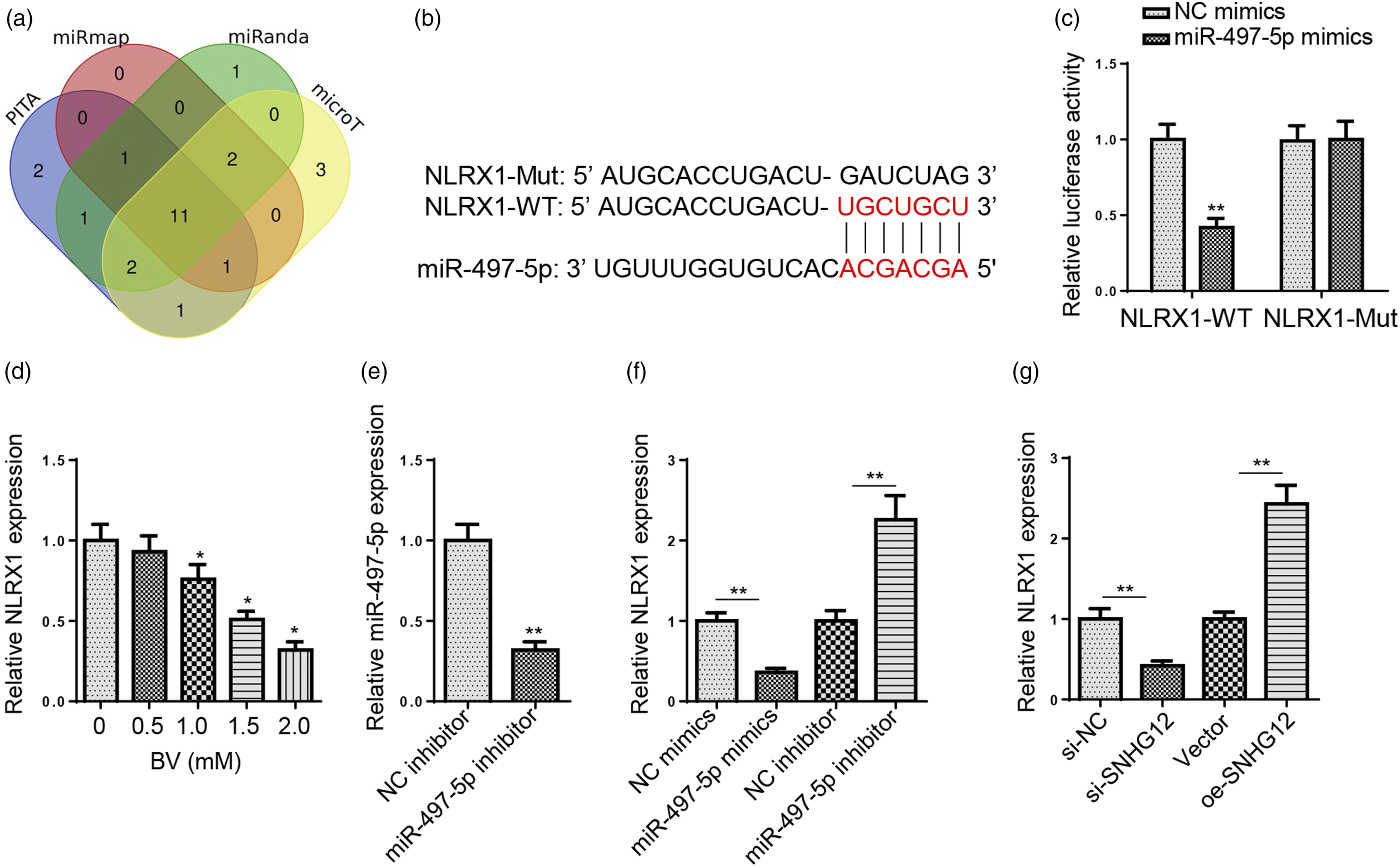
SNHG12/miR-497–5p/NLRX1 axis in BV-induced neurotoxicity
To further explore whether NLRX1 was a downstream regulator of SNHG12/miR-497–5p axis in BV-induced neurotoxicity, SH-SY5Y cells were subject to BV treatment and then transfected with NC inhibitor, miR-497–5p inhibitor, miR-497–5p inhibitor+si-NC, or miR-497–5p inhibitor+si-NLRX1. Firstly, NLRX1 knockdown efficiency in SH-SY5Y cells was confirmed by RT-qPCR (Figure 6(A)). As indicated in RT-qPCR results, miR-497–5p knockdown upregulated NLRX1 expression, which was partly offset by NLRX1 inhibition (Figure 6(B)). Moreover, rescue assays revealed that miR-497–5p downregulation protected SH-SY5Y cells from BV-induced neurotoxicity by increasing cell viability, decreasing cell apoptosis, relieving oxidative damage, and increasing BDNF level; whereas NLRX1 depletion partially reversed these effects (Figure 6(C–I)). In addition, SNHG12 overexpression markedly overturned the suppressive effect of miR-497–5p addition on NLRX1 expression in BV-stimulated SH-SY5Y cells (Figure 6(J)). Taken together, these results indicated that SNHG12 ameliorated BV-induced neurotoxicity via the miR-497–5p/NLRX1 axis.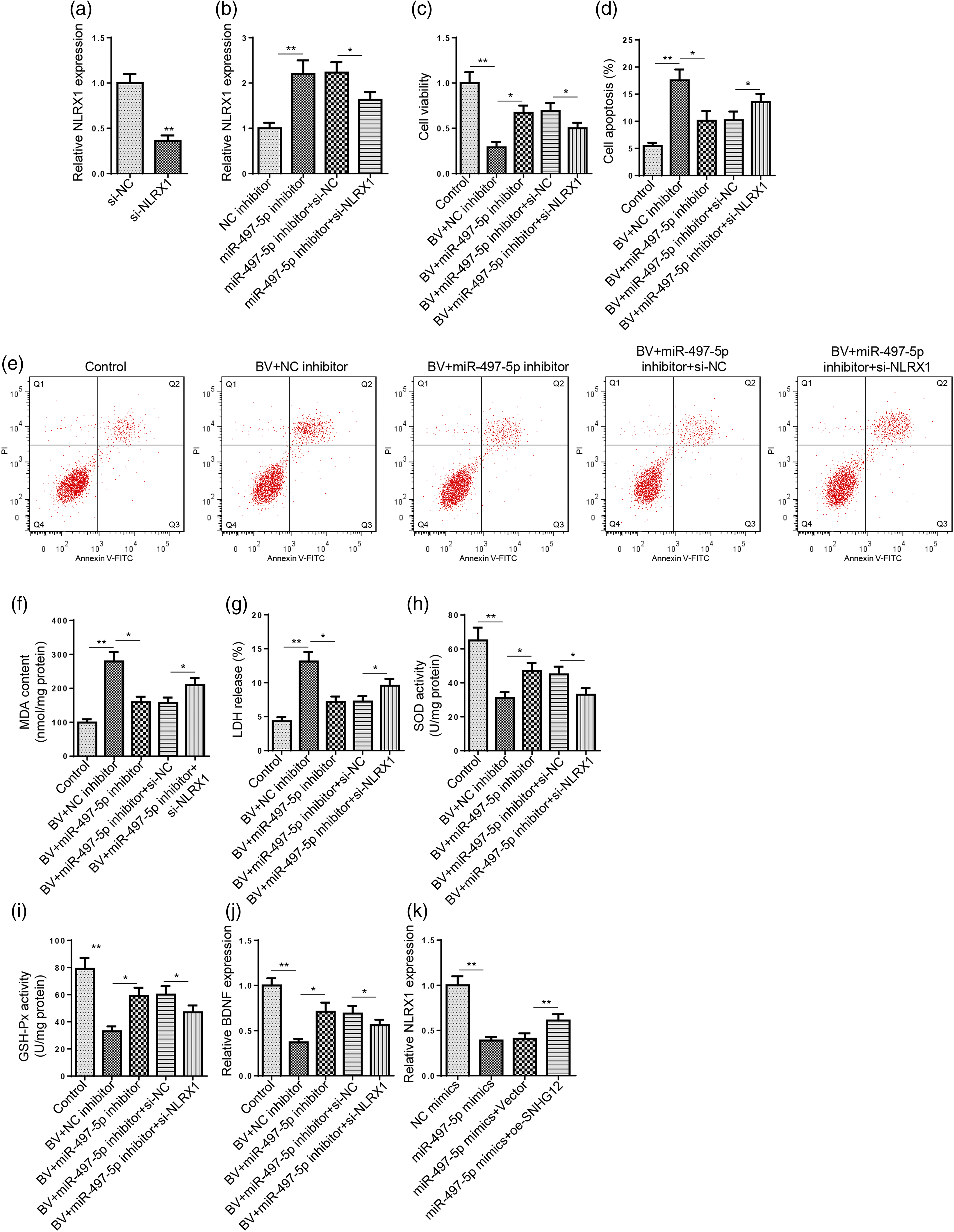
Discussion
BV could cause a lot of adverse drug reactions of which neurotoxicity might lead to permanent and irreversible neurological complications.
26
Increasing evidence indicates that lncRNAs play vital roles in BV-induced neurotoxicity. Figure 7 For instance, Fan et al. demonstrated that lincRNA PADNA could alleviate BV-induced neuronal damage by targeting FBXW7 via miR-194.
2
Guan et al. found that SNHG16 attenuated BV-induced neurotoxicity by negatively regulating miR-132–3p.
27
Yuan et al. revealed that ZFAS1 attenuated BV-induced neurotoxicity via the miR-421/ZNF564 pathway.
28
Numerous studies have elaborated the essential functions of SNHG12 in human diseases, including atherosclerosis,
29
cerebral I/R injury,
30
and I/R-induced cardiac dysfunction.
31
Besides, Wu et al. revealed that SNHG12 could relieve cerebral I/R injury by activating SIRT1/FOXO3a signaling through inhibition of autophagy and oxidative stress.
32
In addition, Yuan et al. disclosed that SNHG12 promoted cell proliferation and reduced cell apoptosis in breast cancer via regulating the miR-15a-5/SALL4 cascade.
33
In this work, it was observed that SNHG12 expression in SH-SY5Y cells was decreased with the increase of BV concentration. Consistent with the above studies, SNHG12 upregulation remarkably promoted cell viability, reduced cell apoptosis, and attenuated oxidative damage in BV-treated SH-SY5Y cells. Therefore, it was assumed that SNHG12 could ameliorate BV-induced neurotoxicity in SH-SY5Y cells. Schematic diagram shows regulatory mechanisms of SNHG12/miR-497–5p/NLRX1 axis in BV-induced neurotoxicity.
It has been widely recognized that a lncRNA could regulate messenger RNA (mRNA) expression by adsorbing specific miRNA as a competing endogenous RNA (ceRNA). 34 Also, the lncRNA/miRNA/mRNA network is deeply involved in neurotoxicity induced by anesthetics, including BV.23,35 Former studies have proven the promoting role of miR-497–5p in neuronal apoptosis induced by MPP+ and ketamine (an anesthetic).24,36 Herein, it was found that miR-497–5p level in SH-SY5Y cells was upregulated by BV treatment in a dose-dependent manner. In addition, further investigation demonstrated that SNHG12 negatively regulated miR-497–5p and alleviated BV-induced neuronal damage in SH-SY5Y cells through interaction with miR-497–5p. The above results indicated that SNHG12 modulated BV-induced neurotoxicity in vitro via targeting miR-497–5p.
NLRX1 is a member of the nucleotide-binding oligomerization domain, leucine-rich repeat containing protein (NLR) family, 37 and uniquely localized to the mitochondria. 38 As a negative regulator of inflammatory responses, NLRX1 can negatively regulate inflammatory cytokine production and is deeply involved in the regulation of neuroinflammation.39,40 As demonstrated by several studies, NLRX1 exerts neuroprotective effects against traumatic brain injury25,41 and neurodegeneration. 42 Moreover, Arab et al. revealed that NLRX1 could attenuate oxidative stress and apoptosis via regulating mitochondrial activity. 43 Li et al. demonstrated that NLRX1 could alleviate apoptosis and inflammatory responses in myocardial ischemia. 44 Also, NLRX1 upregulation could substantially improve the viability of lipopolysaccharide-challenged chondrocytes. 45 In this study, we found that NLRX1 level in SH-SY5Y cells was decreased with the increase of BV concentration. Besides, it was verified that SNHG12 positively regulated NLRX1 expression in BV-treated SH-SY5Y cells via absorbing miR-497–5p. Moreover, NLRX1 knockdown could partially neutralize the effects of miR-497–5p inhibition on BV-mediated cell viability inhibition, cell apoptosis, and oxidative damage in SH-SY5Y cells.
Conclusions
Our investigation demonstrated that SNHG12 relieved BV-induced neurotoxicity in vitro by sponging miR-497–5p to upregulate NLRX1, thereby offering potential molecular targets for the diagnosis and treatment. Nevertheless, the present study lacked in vivo experiments, which should be performed in future studies to further validate the biological functions of the SNHG12/miR-497–5p/NLRX1 regulatory network in BV-induced neurotoxicity.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.