
Abstract
Purpose
Osteosarcoma (OS) is a primary malignant bone tumor, and the cure rate has stagnated in the past three decades. Butein, a plant polyphenol extracted from many herbs, has been proved to possess anti-tumor activity. However, the effect of butein on human OS and the underlying mechanisms remain to be elucidated.
Materials and Methods
The OS cell line 143B was used. The effects of butein were evaluated through the cell proliferation assay, flow cytometry, florescence and transmission electron microscopy, and western blotting. All statistical analyses were performed using GraphPad Prism 7.0.
Results
Butein was found to inhibit cell proliferation by causing G2/ M phase arrest in the 143B cells. In addition, butein suppressed the invasion of 143B cells upon IL‐6 treatment. Additionally, we found that butein inhibited the invasion of 143B cells stimulated with IL-6 via the p-STAT3-MMP9 signaling pathway. Remarkably, butein triggered extrinsic and intrinsic apoptosis and autophagy of 143B cells. The process of autophagy may have tumor-supporting effects. Furthermore, butein induced oxidative stress as evidenced by ROS generation, increase in malondialdehyde (MDA) level, and decrease in GSH/GSSH ratio and GPX4 expression. N-acetylcysteine can reverse the change of ROS. Further experiments indicated apoptosis and autophagy could be attenuated by the N-acetyl-L-cysteine and c-Jun N-terminal kinase (JNK) inhibitor SP600125. Additionally, butein inhibited the Akt/mammalian target of rapamycin (mTOR) signaling pathway, and suppressed the Akt kinase activity increased apoptosis and autophagy.
Conclusion
Our results revealed butein induced apoptosis and autophagy by regulating oxidative stress, activating the JNK signaling pathway and blocking the Akt/mTOR signaling pathway in OS cells. Additionally, butein inhibited the invasion of 143B cells stimulated with IL-6 through the pSTAT3- MMP9 signaling pathway. In view of these results, butein may be a potential anti-tumor drug targeting osteosarcoma.
Introduction
Osteosarcoma is the most common primary bone malignancy in children and adolescents that emerges principally in the metaphysis of long bones with abundant blood supply and is characterized by early metastasis, poor prognosis, and high mortality. 1 Approximately 2–3 and 8–11 patients per million adults and 15–19-year-old individuals, respectively, are diagnosed with OS annually. 2 Owing to the advancement of surgical method and the development of multi drug and dose-intensive chemotherapy, the overall 5-year survival rate of OS has increased from < 20% to 70% within the last two decades. Nevertheless, the 5-years survival rate of patients with early lung metastasis is <20%and has not substantially changed.3,4 In the past three decades, no significant breakthrough has been witnessed in the treatment of OS, and the overall survival rates have plateaued with multi-agent chemotherapy. Moreover, chemotherapy resistance and systemic toxicity of chemotherapy drugs limit the application of these drugs. 5 Therefore, developing novel natural products with high efficacy and less side effects are highly warranted.
Butein(2’,3,4,4’-tetrahydroxychalcone), is a chalcone polyphenol that can be extracted from many herbs, such as Rhus verniciflua Stokes, Caragana jubata, Toxicodendron vernicifluum, and Dalbergia odorifera. Similar to other natural chalcone substances, butein exhibits many biological activities, such as antioxidant, anti-inflammatory, antidiabetic, antibacterial, and neuroprotective effects. Studies have demonstrated its great potential in the treatment of various diseases, such as cancer, inflammatory diseases, infectious diseases, liver diseases, obesity, diabetes, and neuropathy.6,7 Recent studies have reported that the potential antitumor activity of butein in various malignancies, including lung cancer, gastric cancer, and breast cancer.6-10 However, whether butein suppresses OS development and the underlying molecular mechanisms remains to be explored.
Oxidative stress is a negative effect produced by free radicals in the body, and is considered to be an important factor leading to aging and diseases. Reactive oxygen species (ROS) are produced under oxidative stress, and exert substantial effects on normal cell growth and pathological processes. 11 ROS could show beneficial effects at low levels. However, the excessive accumulation of ROS can promote cancer occurrence and development. 12 Numerous studies have demonstrated the role of ROS in apoptosis and autophagy of tumor cells. ROS can activate various signaling pathways, such as JNK, a stress-activated protein kinase in the mitogen-activated protein kinase family, which plays an important role in many cellular events, including apoptosis and autophagy. 13 In addition, the Akt/mammalian target of rapamycin (mTOR) signaling pathway is considered to be one of the main regulatory pathways for cell survival in both normal cells and cancer cells.14,15 Many antitumor drugs have been proved to activate apoptosis and autophagy by inhibiting the AKT/mTOR pathway, which is also an important signal pathway affected by ROS.16,17
In the current study, we investigated the anti-tumor effects of butein on OS. Furthermore, we discuss the possible mechanisms of the crosstalk between apoptosis and autophagy that is mediated through oxidative stress-induced JNK activation and mTOR inhibition.
Materials and methods
Cell and cell culture
The human OS cell line 143B was obtained from Zhongqiaoxinzhou Biotech (Shanghai, China). The cells were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) 100 U/mL penicillin and 100μg/mL streptomycin at 37°C with 5% CO2.
Reagents and antibodies
Butein (purity of > 98%) was purchased from Shanghai Yuanye Biotechnology, Ltd. (Shanghai, China). Stock solution at 100 mM was made in DMSO (Sigma, St. Louis, MO, USA) and stored in the dark at −20°C. 3-Methyladenine (3-MA), broad-spectrum caspase inhibitor z-VAD-fmk-VAD(VAD), SP600125(SP), MK2206(MK), and N-acetyl-L-cysteine (NAC) were purchased from Target Mol. Hoechst staining kit, terminal deoxynucleotidyl transferase -mediated dUTP Nick- End Labeling (TUNEL) kit and JC-1 kit were bought from Beyotime Institute of Biotechnology (Jiangsu, China). The antibodies to GPX4, LC3B, phospho-AKT, β-actin and GAPDH were purchased from Proteintech (Chicago, IL, USA). The antibody to p-mTOR and p-JNK were bought from Cell Signaling Technology.
Cell proliferation assay
The cells were incubated overnight in a 96-well plate and then treated with various concentrations of 0,6.25,12.5, 25,50, and 75μM for 24 and 48 h. Cell viability was assessed using cell counting kit-8 (CCK8) (Dojindo, Kumamoto, Japan) according to the protocol. In brief, the cells were washed with phosphate buffer saline (PBS) and incubated with CCK8 working solution at 37°C for 1–2 h. To determine cell viability, the optical density (OD) at 450 nm was measured using a microplate reader (Tecan infinite M1000 Pro).
Colony formation assay
To examine the capacity of single cells to form a colony, the 143B cells were seeded in 6-well plates at a density of 500 cells/well. After 24 h, the cells were treated with various concentration of 5,7.5, and10 μM for about 14 days. Then, the cells were washed twice with PBS, fixed with 4% paraformaldehyde, and then stained with 0.2% crystal violet solution for 15 min. The clones with>50 cells were counted under a light microscope (ZEISS, Germany), and the images were captured using a camera.
Cell cycle analysis by flow cytometry
The role of butein in cell cycle distribution was analyzed using the propidium iodide (PI)/RNase staining buffer. Briefly, the cells were incubated in 6-well plates and treated with butein or DMSO for 24 h. Then, the cells were harvested, washed twice with ice-cold PBS, and fixed overnight with precooled 75% ethanol at 4°C. Next, the cells were incubated with 500 μL of PI/RNase A staining buffer (PI/RNase A at 9:1) in the dark for 30 min at RT. After staining, the samples were evaluated using the flow cytometer (FACSCalibur, BD, San Jose, CA, USA) and FlowJo™ software.
Transwell invasion assay
The cells that were either pretreated or not treated with butein and IL-6 for 24h were collected and re-suspended in culture medium. Afterward, the upper chambers coated with Matrigel matrix (BD, USA) were filled with 200 μL of cell suspension. Then, the upper chambers were placed into a 24-well plate that containing 12% FBS-supplemented DMEM (600 μl). After incubation for 24h, the cells were fixed with 4% polyformaldehyde, and stained with 0.1% crystal violet for 30 min. The cells at the upper surface were softly wiped off using a cotton swab. Images were captured under an inverted microscope (ZEISS, Germany) and then the cell number was calculated.
Morphological apoptosis
Hoechst 33342 staining was conducted to observe apoptosis. The 143B cells were inoculated on the coverslips in 6-well plates overnight and treated with butein for 24 h. After treatment, the Hoechst 33342 solution was used to analyze morphological changes in the nucleus according to the manufacturer's instructions (Beyotime, China). Finally, the cells were observed under a fluorescence microscope (ZEISS, Germany).
TUNEL assay
The 143B cells were plated on coverslips in 24-well plates and treated with butein for 24 h. After 24 h, apoptosis in the 143B cells was examined using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay kit (Beyotime, China) according to the manufacturer’s instructions. In brief, cells were fixed with 4% paraformaldehyde for 30 min at room temperature and permeabilized with 0.3% Triton X-100 for 5 min at RT. Then, the cells were incubated with 50 μL of TUNEL test solution in the dark at 37 ° C for 1 hour. The nuclei were stained with Hoechst staining solution (Beyotime, China). Finally, the TUNEL-positive cells were examined under a fluorescence microscopy (ZEISS, Germany).
Measurement of mitochondrial membrane potential (MMP)
The MMP was measured using the JC-1 Assay Kit (Beyotime, China) according to the manufacturer's instructions. Briefly, 143B cells were seeded in 6-well plates and treated with butein for 24h. After treatment, the cells were incubated with 1mL of JC-1 solution for 20 min at 37°C. The cells were observed under a fluorescence microscope (ZEISS, Germany), and mitochondrial depolarization was indicated by a decrease in the red/green fluorescence intensity ratio.
Flow cytometric analyses of apoptosis
The 143B cells were sub-cultured in 6-well plates and treated with butein at indicated concentration for 24 h. Then, the cells were collected, washed twice with chilled PBS, and then resuspended in the 1X binding buffer. Aliquots of 105 cells were stained with 5 μL of APC Annexin V and 5 μL of PI for 15 min at RT in the dark. After staining, the samples were evaluated using the flow cytometer (FACSCalibur, BD, San Jose, CA, USA) and FlowJo™ software.
Measuring apoptotic activity
The 143B cells were treated with butein with or without NAC. Afterward, the cells were collected and lysed on ice with lysis buffer (Beyotime, China) and centrifuged at 20000×g for 15 min at 4°C. The supernatants were collected to detect the activities of caspase-8, -9, and -3 by using colorimetric assay kits according to the manufacturer’s instructions. The caspase activity was evaluated by measuring the absorbance at 405 nm. The results were then standardized to the protein concentrations.
Oxidative stress detection
To determine whether butein induced oxidative stress in the 143B cells, we measured the levels of ROS, MDA, GSH/GSSG ratio, and GPX4 protein expression level. The production of ROS was detected by using the ROS Assay Kit (Beyotime, China). In short, the cells were seeded in 6-well plates and treated with butein for 24 h. The cells were stained with 10 μM DCFH-DA at 37°C in the dark for 30 min. Then, the cells were washed thrice with serum-free DMEM and the ROS level was measured using a fluorescence microscopy (ZEISS, Germany). The lipid peroxidation assay was conducted using the MDA assay kit. Briefly, the 143B cells were treated with butein. Then, the cells were collected and lysed with lysis buffer and then centrifuged at 12000×g for 10 min. Subsequently, the cell supernatants were collected and added with the test solution. Then, the EP tubes containing the samples and test solution were kept in boiling water for 15 min and cooled to RT. Finally, the MDA level was evaluated by measuring the absorbance at 532 nm and standardized to the protein concentrations. The GSH/GSSG assay kit was used to detect the levels of total glutathione and oxidized glutathione. The 143B cells were treated with butein for 24h. Next, the cells were washed with PBS. Then, the cell samples were subjected to rapid freezing and thawing twice with liquid nitrogen and 37°C water bath. After centrifugation at 10000×g for 10 min, the cell supernatants were harvested and processed with the test solution. Finally, the GSH level was evaluated by measuring the absorbance at 412 nm. Total glutathione minus GSSG represents the GSH level.
Western blot assay
The cells were seeded in 100-mm dishes and treated with butein and SP or MK for 24 h. The 143B cells samples were lysed in cooled RIPA buffer containing protease inhibitor mixture (Beyotime, China) for 30 min. Then, the lysates were centrifuged at 12,000g and4 °C for 15 min and the supernatant was collected. The protein concentration was quantified using the BCA protein assay kit (Beyotime biotechnologies, Jiangsu, China) according to the manufacturer’s instruction. Equal amounts of protein were separated by SDS-PAGE and transferred to the polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk for 1 h at room temperature (RT) and then incubated with primary antibodies overnight at 4°C, subsequently incubated with species-specific peroxidase-conjugated secondary antibodies for 1 h at RT. Each band was detected by using a chemiluminescence reagent and visualized using a Bio-Rad imaging system.
Transmission electron microscopy
The 143B cells were treated with butein for 24 h. The treated cells were fixed with 2.5% glutaraldehyde for 1 h at RT and post-fixed with 1% osmium tetroxide for another 1 h. The cell pellets were dehydrated in graded ethanol series, and then embedded in epon. Representative areas were chosen for ultrathin sectioning. After making ultrathin sections, the sections were stained with uranyl acetate and lead citrate. The morphology of the autophagosomes was examined under a transmission electron microscope. (H-7650, Hitachi, Tokyo, Japan).
Statistical analysis
The quantitative data are presented as the mean ± SD. Fisher’s Exact test, unpaired student's t-test, one-way ANOVA and two-way ANOVA were used to compare differences between the groups. All statistical analyses were performed using GraphPad Prism 7.0 (San Diego, CA, USA). p-values < 0.05 were considered as statistically significant.
Results
Butein inhibited 143B cells viability
The CCK8 assay and colony formation test were used to examine the anti-proliferative effect of butein on the 143B cells. As shown in (Figure 1(a)), cell proliferation activity was significantly decreased in the dose- and time-dependent manner. The IC50 for 143B cells at 24h and 48h were 23.9 μM and 11.7 μM, respectively. Additionally, the colony formation test demonstrated that butein treatment significantly inhibited colony formation (Figure 1(b)). These results suggested that butein inhibited 143B cells proliferation. Butein inhibited cell viability and induced G2/M cell cycle arrest in human 143B cells. (a) Cells were treated with butein (0, 6.25, 12.5, 25, 50 and 75 μM) for 24 and 48 h, cell viability was measured by CCK8 method. (b) The ability of cell colony formation was evaluated by clonogenic assay. (c) Butein induced G2/M cell cycle arrest in 143B cells. Cells were treated with butein 24 h and then analyzed by flow cytometry. *p <0.05, significantly different compared with the untreated control group. CCK8: cell counting kit-8.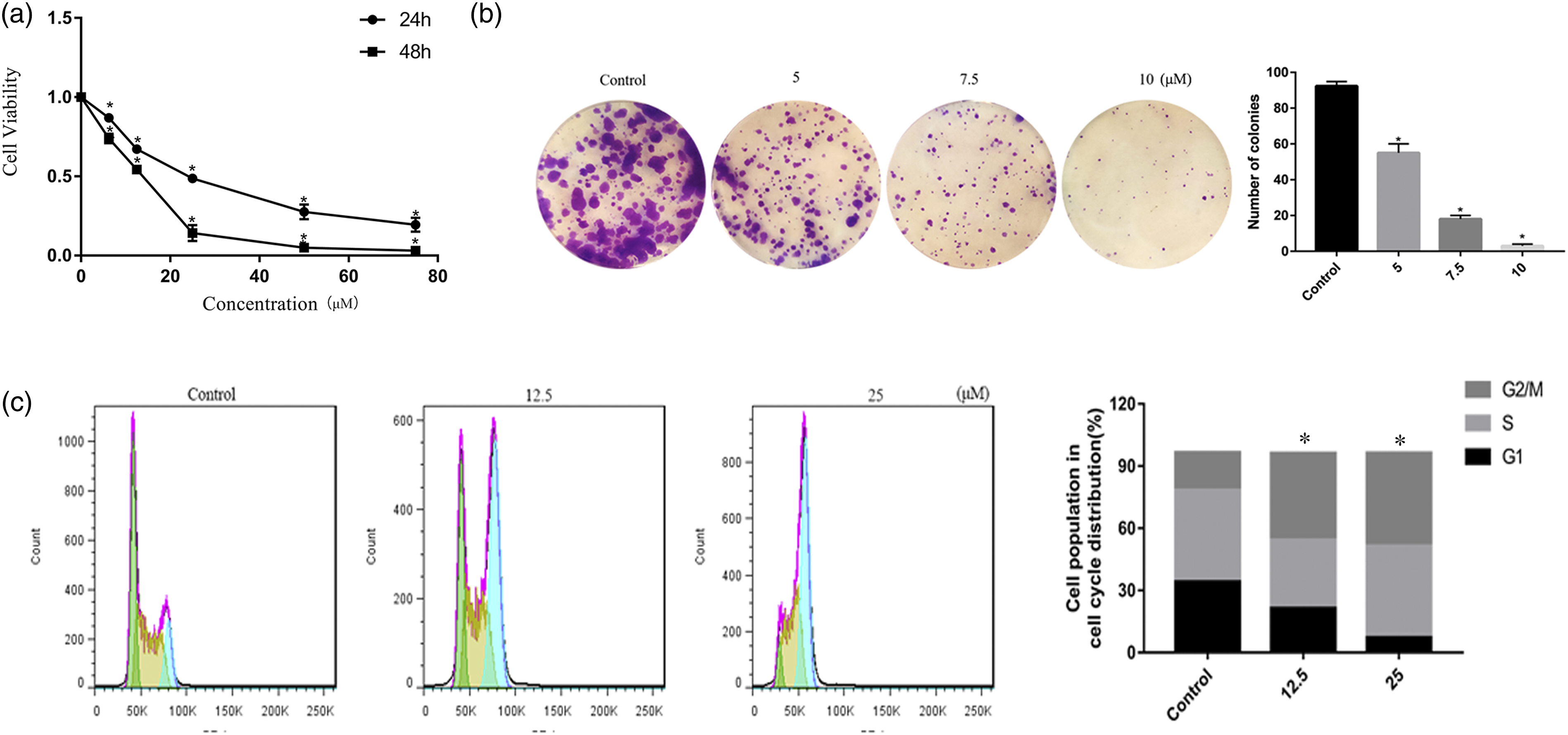
Butein induced cell cycle arrest at G2/M phase in 143B cells
To determine the relationship between growth inhibition and cell cycle arrest, we examined the effect of butein on cell cycle progression. As shown in Figure 1(c), butein treatment led to the accumulation of cells in the G2/M phase, accompanied by G0/G1 phase and S phase reduction. These results indicated butein inhibited 143B cell growth by inducing cell cycle arrest at the G2/M phase.
Butein inhibits the invasiveness of 143B cells stimulated by IL-6
Because 143B cells are highly aggressive OS cells, and inflammatory factors in cancer often promotes tumor cell invasion. Next, we investigated the effect of butein on the invasiveness of 143B cells stimulated by IL-6. Transwell invasion test showed that butein significantly inhibited the invasion of 143B cells upon IL-6 treatment (Figure 2(a)). We further investigated the effect of butein on E-cadherin expression, which indicated that butein upregulated E-cadherin expression in 143B cells upon IL-6 treatment. In addition, butein decreased MMP-9 and MMP2 protein expression in the presence of IL-6. IL-6 can increase the phosphorylation of STAT3, and the increase of STAT3 phosphorylation level can promote cancer cell metastasis. Thus, we investigated the effect of butein on the STAT3 signal in the IL-6-stimulated 143B cells. Inevitably, butein inhibited STAT3 phosphorylation (Figure 2(b)). These results indicated that butein can effectively inhibit the invasion of 143B cells. Butein represses the invasion of 143B cells stimulated with IL-6. (a) Butein inhibited the invasion ability of 143B cells stimulated with IL-6. (b) The expression levels of E-Cadherin, MMP9, MMP2, and p-STAT3 were detected by Western blotting. *p<0.05 versus control, #p<0.05. MMP: mitochondrial membrane potential.
Butein induced apoptosis of 143B cells
Butein-induced apoptosis was investigated through Hoechst 33258 staining, TUNEL assay, MMP assay, and Annexin V/PI assay. As shown in Figure 3(a), the 143B cells treated with butein for 24 h showed some apoptotic characteristics as indicated by the red arrows, such as chromatin condensation, and nuclear fragmentation. TUNEL staining results further revealed that butein treatment led to an increase in DNA fragmentation in the 143B cells (Figure 3(b)). MMP (ΔΨm) loss is a crucial step in the process of apoptosis. Next, we analyzed it by using JC-1 probe and found a decrease in the red/green fluorescence intensity ratio following butein treatment (Figure 3(c)). Additionally, we quantified apoptosis through Annexin V-PE/APC double staining. The results demonstrated that the exposure to 12.5µM–50μM butein lead to a dose-dependent increase in the number of apoptotic cells (Figure 3(d)) To further verify that butein induced apoptosis of 143B cell, we determined the activities of caspsase-3, -8, and -9. As shown in Figure 3(e), the caspase-3 activity was significantly increased in the 143B cells treated with butein (p < 0.05). The activity of caspase 8 (an extrinsic apoptotic pathway initiator caspase) and caspase 9 (an intrinsic apoptotic pathway initiator caspase) was also significantly increased in a dose-dependent manner (p < 0.05), which indicated that butein triggered both extrinsic and intrinsic apoptosis in the 143B cells. Moreover, VAD (a caspase inhibitor) could reverse a certain proportion of cell death (Figure 3(f)). Butein induced apoptosis in 143B cells. (a) The morphological changes of apoptotic nuclei were evaluated by Hoechst 33258 staining and observed under a fluorescent microscopy. Chromatin condensation and DNA fragmentation was indicated by red arrows. Scale bars: 50 μm. (b) 143B cells treated with butein were stained with Tunel kit and evaluated by a fluorescent microscopy. Scale bars = 50 μm. (c) The MMP was measured with JC-1 fluorescent probe and assessed by a fluorescent microscopy. Scale bars = 20 μm. (d)143B cells treated with butein were stained with annexin V-APC/PI and evaluated by flow cytometry. (e). The changes of caspase-3,8,9 activities after butein treatment. (f)143B Cells were pretreated with caspase inhibitor, VAD and then incubated with control or butein for 24 h. The induction apoptosis was analyzed by flow cytometry. *p<0.05 versus control, #p<0.05 versus butein treatment. MMP: mitochondrial membrane potential; VAD: .
Butein triggered autophagy, which protects cell survival
To determine whether butein could induce autophagy in the OS cells. We first used LysoTracker Red to label the cellular acidic compartments, such as lysosomes and autolysosomes. Figure 4(a) shows that butein-treated 143B cells exhibited more acidic vesicular organelles (AVOs) in the cytoplasm. Furthermore, we determined LC3B expression by western blotting. As shown in Figure 4(b), butein increased LC3B-II level in the 143B cell in a dose-dependent manner. Finally, we observed autophagosomes formation directly through TEM. The results showed that the number of autophagic vacuoles was significantly increased in the cytoplasm of butein-treated cells compared with that in the control cells (Figure 4(c)). Butein induced autophagy, and inhibited autophagy enhanced butein-induced apoptosis. (a) Cells treated with or without butein for 24 h were collected and stained with 50 nM of LysoTracker Red DND-99. Representative images of indicated cells captured by fluorescent microscopy were shown. Scale bars = 50 μm. (b) Cells were treated with indicated concentrations of butein for 24 h. The level of LC3B was measured by western blot. (c) Transmission electron microscopy was utilized to observe the formation of autophagosome. Red arrows indicate autophagosomes containing intact and degraded cellular debris. Scale bar: 2 μm. (d). Cell viability was evaluated using CCK-8 assay following blockade of autophagy by pharmacological inhibitor 3-MA and butein treatment for 24h and 48h. (e). The apoptotic 143B cells were analyzed by flow cytometry following the treatment with butein in presence or absence of 3-MA for 24h. *p<0.05 versus control, #p<0.05 versus butein treatment. CCK8: cell counting kit-8.
Autophagy plays a dual role in cancer treatment; its role is to protect cell survival or lead to cell death. To determine the effect of butein-induced autophagy, we used 3-MA to block butein-induced autophagy in the OS cells. As shown in Figure 4(d), cell viability was decreased compared with that under butein treatment alone. As shown in Figure 4(e), 3-MA treatment increased the extent of butein -induced cell death. These data suggested that butein mediated autophagy promoted 143B cells survival.
Butein induced oxidative stress, and activated the JNK signaling pathway in human 143B cells
Our result revealed that the MMP decreased sharply in the butein-treated 143B cells. Mitochondria are the main source of ROS inside cells. This led us to assume that butein may trigger the release of ROS from mitochondria. Therefore, the fluorescent probe DCFH-DA was used to investigate whether butein treatment could enhance ROS production. As shown in Figure 5(a), the fluorescence signal was significantly increased in butein-treated 143B cells. Additionally, butein increased the level of lipid peroxidation, as evidenced by an increase in the MDA level (Figure 5(b)). The cellular redox balance is critical to cell fate. GSH is the main cell redox buffer, and the GSH/GSSG ratio is an important indicator of cell redox status. After butein treatment, the ROS level was significant increased. Thus, we believed that the antioxidant system exhibited defects, and thus lacked the ability to scavenge ROS. The results revealed that butein significantly reduced the GSH/GSSG ratio (Figure 5(c)). Additionally, the expression level of GPX4, an enzyme that can effectively reduce lipid peroxide, was decreased (Figure 5(d)). Therefore, we considered that butein induced oxidative stress in human 143B cells by damaging the GSH antioxidant defense of the143B cells. However, the ROS level was significantly reduced when the ROS scavenger NAC was used (Figure 5(e)). Butein induced intracellular ROS generation, activated JNK signaling pathway. (a) The intracellular ROS levels was determined by a fluorescence microscopy using DCFH-DA staining. Bar: 50 μm. (b) Butein increased MDA level of 143B cells. (c) Butein significantly reduced the GSH/GSSG ratio. (d) Levels of GPX4 were assessed by western blot. (e) 143B cells were incubated with butein and pretreated with JNK inhibitor (SP600125) or ROS scavenger (NAC). Changes in ROS levels was determined by a fluorescence microscopy. (f)143B Cells were treated with various concentrations of butein for 24 h. Levels of p-JNK were assessed by western blot. Data are represented as mean ± S.D. *p<0.05 versus control. ROS: Reactive oxygen species; JNK: c-Jun N-terminal kinase; GSH: Glutathione; NAC: N-acetyl-L-cysteine.
Next, we investigated the effect of butein on the JNK signaling pathway. Our results showed that butein increased the phosphorylation level of JNK in concentration-dependent manner (Figure 5(f)).
To further investigated the roles of the JNK signaling pathway, we used NAC and SP were used. Notably, the CCK8 analysis indicated that NAC and SP significantly attenuated butein-induced cell viability inhibition (Figure 6(a)). Flow cytometric analysis demonstrated that NAC had an obvious inhibitory effect on butein-induced G2/M phase arrest (Figure 6(b)) and cell apoptosis, and SP also had an inhibitory effect on butein-induced apoptosis (Figure 6(c)). NAC is more effective in preventing cell death than SP. The activities of caspsase-3 were also obviously reversed by NAC treatment (Figure 6(d)). Interestingly, NAC pretreatment reduced butein-induced autophagy, which was determined by western blot analysis (Figure 6(e)). Furthermore, NAC blocked JNK phosphorylation (Figure 6(e)), whereas SP did not suppress ROS generation (Figure 5(e)). These results suggested that JNK activation following the enhanced ROS production contributed to butein-induced autophagy and apoptosis. Roles of ROS and JNK in cell apoptosis and autophagy induced by butein. 143B cells were incubated with butein and pretreated with JNK inhibitor (SP600125) or ROS scavenger (NAC). (a) Cell viability of 48h was evaluated by CCK8 assay. (b) Cell cycle of 24h was assessed by flow cytometry. (c) Induction of apoptosis of 24h was assessed by flow cytometry. (d) The change of caspase three activity after butein and NAC treatment. (e) Changes of p-JNK and LC3B of 24h were evaluated by western blot. *p<0.05 versus control, #p<0.05 versus butein treatment. ROS: Reactive oxygen species; JNK: c-Jun N-terminal kinase; NAC: N-acetyl-L-cysteine; CCK8: cell counting kit-8.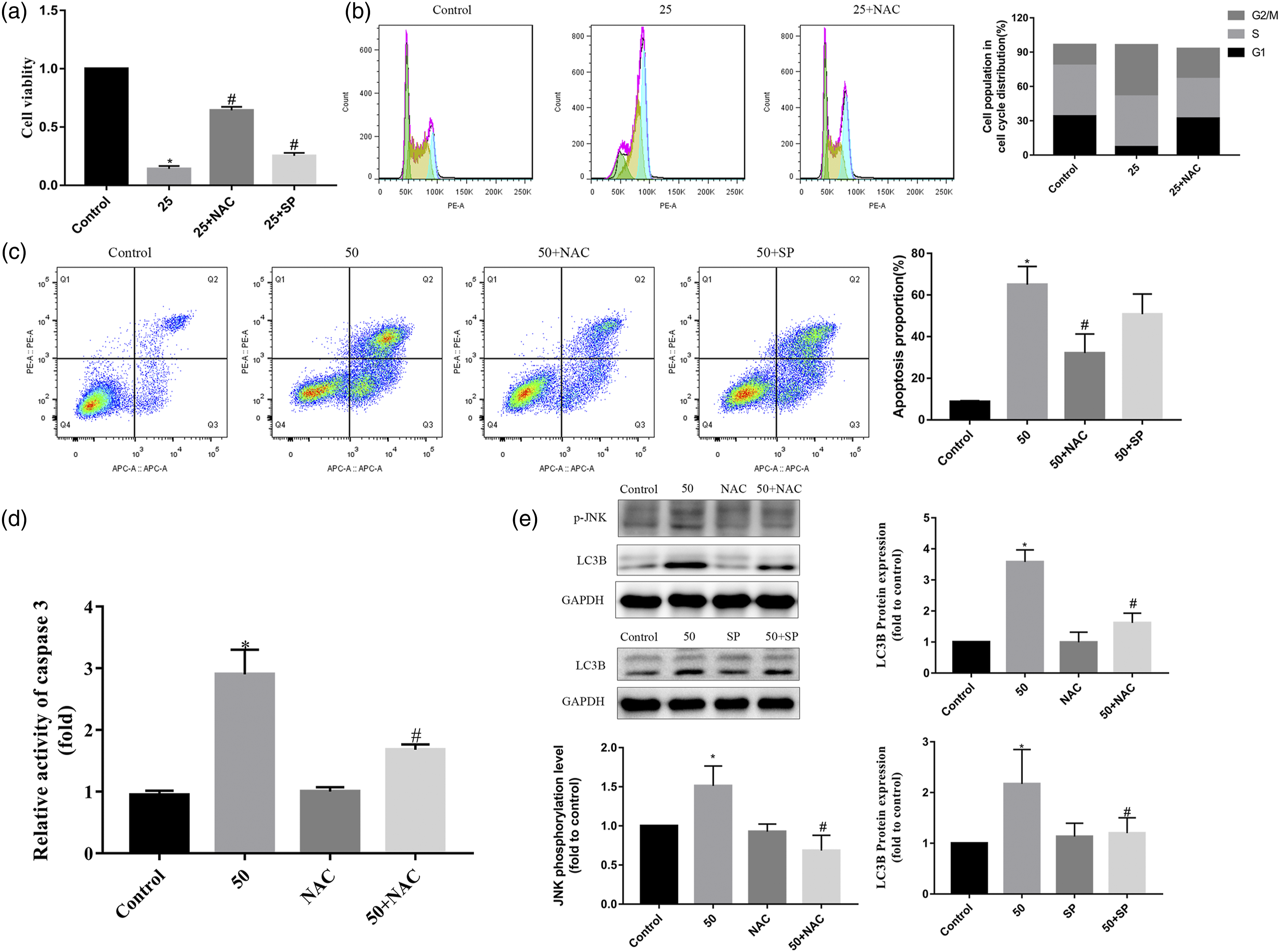
Butein induced apoptosis and autophagy by blocking the Akt/mTOR signaling pathway in human 143B cells
To further understand the molecular mechanism of the antitumor effect of butein, we investigated the effect of butein on the Akt/mTOR signaling pathway. MK (a AKT inhibitor) was used. As shown in Figure 7(a), butein decreased the phosphorylation level of AKT and mTOR. The CCK8 assay exhibited that MK could strengthen the cell inhibitory effect (Figure 7(b)). Flow cytometric analysis demonstrated that MK had led to an obvious increase in cell apoptosis (Figure 7(c)). Additionally, pretreatment of OS cells with MK increased butein induced autophagy, which was determined by the western blot analysis. (Figure 7(d)) Furthermore, NAC increased the phosphorylation levels of Akt and mTOR (Figure 7(e)). These results suggest that butein could induce apoptosis and autophagy through the ROS-Akt/mTOR signaling pathway in human 143B cells. Effect of the Akt/mTOR signaling pathway on butein-induced apoptosis and autophagy. 143B cells were incubated with butein and pretreated with MK2206 (a Akt inhibitor). (a) 143B Cells were treated with various concentrations of butein for 24 h. Levels of p-Akt, and p-mTOR were assessed by western blot. (b) Cell viability of 24 h was measured by CCK8 assay. (c) Cell apoptosis of 24h was evaluated by flow cytometry. (d and e) Levels of LC3B, and p-Akt, p-mTOR were analyzed by western blotting. *p<0.05 versus control, #p<0.05 versus butein treatment. mTOR: mammalian target of rapamycin; CCK8: cell counting kit-8.
Discussion
Butein is a natural product that is isolated from many herbs. Many studies on the antitumor effect of butein have shown that it can effectively inhibit cancer cell proliferation, invasion, and metastasis and induce cell apoptosis in vitro and in vivo.8-10 However, its effectiveness and potential antitumor mechanism in OS treatment have not been fully clarified. In the present study, we observed that butein could inhibit cell proliferation and invasion, trigger G2/M phase arrest, and induce apoptosis and autophagy in human 143B cells. More importantly, butein was found to induce apoptosis and autophagy by regulating oxidative stress, activating the JNK signaling pathway and blocking the Akt/mTOR signaling pathway.
Cell proliferation occurs under an effectively controlled cell cycle, which is regulated by cyclin dependent kinase and CDK inhibitor. The G2/M checkpoint is a key factor for cell proliferation and is considered as one of the significant targets of anticancer drugs. 18 Many cytotoxic drugs have been found to play a crucial role in G2/M cell cycle arrest. 19 The flow cytometry analysis indicated that butein could induce G2/M phase arrest in the 143B cells. Cell cycle arrest could induce cell apoptosis. Apoptosis plays a key role in regulating tumor formation and therapeutic response. The current tumor treatment mainly involves the activation of the apoptosis signal pathway. 20 Our results showed that the MMP decreased significantly after butein treatment, indicating that mitochondrial depolarization was induced by butein in 143B cells. Mitochondrial membrane depolarization can lead to the release of cytochrome c from the space within the mitochondrial membrane to the cytoplasm and activate cytoplasmic caspase. 21 In the current study, butein induced apoptosis in 143B cells, which was confirmed through Hoechst 33258 staining, TUNEL assay, MMP assay, Annexin V/PI assay and apoptotic activity assay. The activities of caspase-3, -8, and -9 were significantly increased after butein treatment, which indicated that butein triggered both extrinsic and intrinsic apoptosis in 143B cells. In addition, VAD treatment significantly reduced butein induced cell death.
Autophagy is considered as a conserved intracellular degradation system, which can degrade and recycle damaged or unnecessary cytoplasmic contents under stress. 22 It plays an important role in many physiological and pathophysiological processes.22-24 A large amount of evidence has revealed that autophagy has dual roles in cancer progression and inhibition. Autophagy can sometimes prevent apoptosis by promoting the survival of cancer cells, and sometimes induce and promote cancer cell apoptosis. 25 The present study demonstrated autophagy induced by butein on the basis of more AVO in the cytoplasm, up regulation of LC3B-II protein expression and the formation of autophagosomes observed through TEM. The study also revealed that the autophagy inhibitor 3-MA could strengthen the inhibition of cell viability and the ability of butein to induce apoptosis, suggesting that butein-induced autophagy may contribute to cell survival.
Oxidative stress is considered to be one of the important mechanisms of anticancer agents to promote cancer cell death. In the present study, butein induced a significant increase in ROS generation and the degree of lipid peroxidation. Furthermore, the GSH/GSSG ratio and GPX4 expression level were decreased, which indicated that butein damaged the GSH antioxidant defense of 143B cells. All of these lead to the oxidative stress of 143B cells. ROS are important upstream molecules that regulates cancer cell death and survival. The change in the ROS level plays an important role in tumorigenesis and is considered to be a promising tumor treatment strategy.26,27 However, pretreatment with NAC remarkably reversed butein-induced ROS generation. JNK and AKT/mTOR are a part of many downstream cascades of the ROS signaling pathway, which could transduce oxidative stress signals and promote apoptosis and autophagy.28,29 In this study, butein treatment triggered JNK activation and mTOR inhibition. We further studied the effects of JNK and Akt/mTOR signaling pathways on butein-induced apoptosis and autophagy. The results showed that the inhibition of JNK activation with SP could reduce butein-induced autophagy and apoptosis. However, MK produced the reverse effects. However, pretreatment with NAC remarkably reversed butein-induced effects such as cell proliferation inhibition, G2/M phase arrest, apoptosis, autophagy, JNK activation, and mTOR inhibition.
Inflammation in cancer microenvironment can actively promote cancer cell migration and invasion. Therefore, the well-known proinflammatory factor IL-6 was used to determine the effect of butein on the invasion of 143B cells. IL-6 can strongly stimulate the activation of STAT3 and play a vital role in the proliferation and invasion of tumor cells. It can also interact with TGF- β or other cytokines to induce epithelial–mesenchymal transition, thereby promoting tumor proliferation, movement, and invasion.30,31 In our study, we found that butein inhibited the invasion of 143B cells upon IL-6 treatment, upregulated the expression of E-cadherin, and downregulated the expression of MMP9 and MMP2 in 143B cells stimulated by IL-6. Additionally, STAT3 phosphorylation were was reduced by butein in IL-6-stimulated 143B cells. Taken together, butein inhibited the invasion of 143B cells stimulated with IL-6 via the p-STAT3-MMP9 signaling pathway.
In conclusion, the present study reports for the first time that butein induced apoptosis and autophagy by regulating oxidative stress, activating the JNK signaling pathway and blocking the Akt/mTOR signaling pathway in OS cells. Additionally, butein inhibited the invasion of 143B cells stimulated with IL-6 through the pSTAT3- MMP9 mediated signaling pathway. This study not only further understood the potential molecular mechanism of butein, but also provided an alternative strategy for OS treatment by using the combination of butein and autophagy inhibitors.
Footnotes
Author Contributions
Wang JC, Zhang P, and Liang Y contributed to the conception and design of the study. Zhang P, Zhang JL, Quan HH performed the experiments. Zhang P and Chen PJ analyzed the data. Zhang P drafted the manuscript. All authors contributed to the manuscript writing and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (No: 81772332); Jiangsu medical innovation team project (No: CXTDB2017004) in analysis and interpretation of data.