
Abstract
This study aims to explore the effect of NDRG2 (N-myc downstream regulated gene 2)-mediated Transforming growth factor-beta 1 (TGF-β1)/ Sma- and Mad-related protein (Smad) pathway in heart failure (HF) rats. HF rat models were established and treated with AdEGFP (adenovirus encoding enhanced green fluorescent protein) or AdNDRG2 (adenovirus encoding NDRG2). The echocardiography and hemodynamic parameters were detected, and the infarct size was calculated via 2,3,5-triphenyltetrazolium chloride (TTC) staining. Masson staining was performed to observe the collagen volume fraction (CVF), quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) to detect the expression of Collagen I (Col-I) and Collagen III (Col-III), and Transferase (TdT)-mediated D-UTP-biotin nick end labeling (TUNEL) staining to evaluate the apoptosis. Rats in the Model group presented with the decreases in left ventricular ejection fraction (LVEF), left ventricular shortening fraction (LVFS), left ventricular systolic pressure (LVSP) and maximal/minimum rate of left ventricular pressure (±dp/dt max), and significant increases in left ventricular end-diastolic pressure (LVEDP) and CVF. At the meantime, the expression of Col-I and Col-III as well as the apoptotic rate of myocardial cells was also elevated with increased infarct size in the Model group. The Model rats also had the significant reduction in the expression of NDRG2 and up-regulations of TGF-β1, p-Smad2/Smad2, p-Smad3/Smad3 and tissue inhibitor of metalloproteinases-2 (TIMP-2). However, model rats treated with AdNDRG2 had evident amelioration in aforementioned indicators. In conclusion, NDRG2 reduces the apoptosis of myocardial cells and improves the heart function and myocardial remodeling in HF rats via inhibiting the activity of TGF-β1/Smad.
Introduction
Heart failure (HF) refers to the clinical syndrome of myocardial damage caused by multiple factors, resulting in the alteration of myocardial structure and function, thus the heart is unable to pump sufficiently to maintain blood flow to meet the body’s needs. 1 HF, as the endpoint event of multiple heart diseases, has been affecting 40 million people globally and severely jeopardizing the health of human beings. 2 Myocardial remodeling is the key factor contributing to the development and progression of chronic heart failure (CHF), presenting with myocardial fibrosis, mesenchymal collagen deposition, disproportionality in the ratios of collagens (mainly the increased ratio of collagen I/III) and disorganized myocardial cells, while the pathological myocardial remodeling further deteriorates the cardiac function, thereby inducing HF. 3 –5 Hence, the work to discover the relevant mechanism of myocardial remodeling in HF shows great clinical significance.
NDRG2, a member of N-myc downstream-regulated gene (NDRG) family, is a kind of tumor suppressor gene closely involved in cell proliferation and differentiation, which is widely distributed with various biological functions. 6 On the basis of the previous literatures, NDRG2 has been shown to participant in the water-salt metabolism, intracellular signal transduction of insulin, as well as the development and aging of nervous system. 7,8 Previous study identified the significantly up-regulated NDRG2 in the myocardial tissues, 9 but the exact role of NDRG2 and its underlying mechanism remain unclear in heart. Sun et al. reported that insulin reduced the myocardial infarction area via inhibiting myocardial cell apoptosis, with up-regulations of Akt and NDRG2 phosphorylation, whereas the administration of shRNA deteriorated the myocardial ischemia-reperfusion injury via down-regulating NDRG2, 10 which provided a possibility that NDRG2 have a pivotal regulatory role in HF. Transformation growth factor-β1 (TGF-β1), a key regulator of the hyperplasia of fibrous connective tissue and synthesis and deposition of collagen, can transduce the signals through the downstream transcription factors, like Smad2 and Smad3, thus playing an important role in myocardial remodeling, but multiple drugs that can delay the myocardial remodeling could also inhibit the TGF-β1/Smad pathway. 11 –13 NDRG2 overexpression, as demonstrated by Yang et al., can suppress the TGF-β1/Smad pathway to promote the degradation of extracellular matrix (ECM), thereby improving the liver function of rats with hepatic fibrosis. 14 Hence, it is reasonable to speculate that NDRG2 may have a role in HF by regulating TGF-β1/Smad.
In line with this hypothesis, HF rat models were established to uncover the role of NDRG2-mediated TGF-β1/Smad pathway in myocardial remodeling and cardiac function of HF rats, aiming to provide an effective therapy for the prophylaxis and treatment of myocardial remodeling after HF.
Materials and methods
Ethical statement
This study was reviewed and approved by the Ethical Committee of our Hospital, and the protocol of animal experiments was in strict accordance with the Guide for the Care and Use of Laboratory Animals published by National Institutes of Health (NIH) in the USA. 15
Subjects and grouping
Male Sprague-Dawley (SD) rats weighing between 200 and 220 g were provided by Shanghai SLAC Laboratory Animal Co., Ltd (China) and kept in specific-pathogen-free (SPF) room at 12 h/12 h light/dark cycle with food and water ad libitum, where the temperature was set at (22 ± 3)°C with the relative humidity at (70 ± 4)%.
Preparation and grouping of the CHF rat models
Prior to the surgery, rats were fasted for 12 h but had free access to the water. Then, rats were anesthetized with 30 mg/kg pentobarbital sodium via intraperitoneal injection, placed in supine position and prepared for the subcutaneous implantation of echocardiography electrodes in the limbs of rats. Thereafter, coronary artery ligation was applied to prepare CHF models on rats (n = 60). In brief, after the thoracic cavity exposed through the incision between the 3rd and 4th rib, the heart was exposed, where the left auricle was lifted slightly using the forceps and, at 2–3 mm beneath the left anterior descending coronary artery, a 6-0 silk suture was used to ligate the left main coronary artery, resulting in myocardial infarction, followed by the closure of the chest. After surgery, standard limb lead and precordial lead were recorded, and any ST segment elevation suggested the successful construction of the model. 2 For rats in the Sham group (n = 12), after operations similar to those in the Model group, coronary artery ligation was substituted by placing a suture under the left anterior descending coronary artery. All of the rats (n = 12) survived from the sham surgery. Two weeks after the coronary artery ligation surgery, 40% (24/60) rats died and the 36 surviving HF rats (the survival rate was 60%) were identified the successful model establishment and then randomly assigned to Model group, Model + adenovirus encoding enhanced green fluorescent protein (AdEGFP) group and Model + AdNDRG2 group with 12 rats in each. Rats in the Model + AdEGFP group and Model + AdNDRG2 group would receive the injection of 4 × 109 PFU AdEGFP and adenovirus encoding NDRG2 (AdNDRG2, Benyuan Zhengyang Inc., Beijing, China) via the tail vein at two weeks after construction of CHF models. 14 After either injection, there was 100% survival in all groups.
Echocardiography
Four weeks after model construction, rats in each group (n = 12) were anesthetized with 30 mg/kg pentobarbital sodium intraperitoneally and conducted echocardiography in supine position. In brief, a GE Vivid 7 ultrasound system (GE, USA) was utilized, with S4 probe at an operating frequency of 2.5 Hz. Firstly, couplant-smeared S4 probe was placed at the left side of sternum and, under the ultrasonic two-dimensional guidance, was used to find out the standard longitudinal section of left ventricle along the left ventricular outflow tract. Then, S4 probe was rotated by 90° to detect the transverse section horizontal to the level of papillary muscles of left ventricle, followed by the two-dimensional ultrasonography and M-type echocardiography to measure the left ventricular ejection fraction (LVEF) and left ventricular shortening fraction (LVFS).
Hemodynamics
Rats were placed in the supine position on the operation table, and an incision was made on the neck, where 1.5 cm of right common carotid artery was isolated. Then, the distal end of this segment was ligated, while the proximal end was clamped by a bulldog clamp. On the arterial wall, a small incision was made, where a polyethylene catheter in inner diameter of 0.5 mm and outer diameter of 1 mm filled with 500 U/L heparin sodium in normal saline was inserted into the left ventricle, and the other end was connected to the physiological recorder (RJG-4122, Nihon Kohden, Japan) via the pressure transducer (TRI 21. Letica Scientific Instruments, Spain). The left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP) and the maximal/minimum rate of left ventricular pressure (±dp/dtmax) were then recorded.
Determination of cardiac hypertrophy and myocardial infarct size
Cardiac hypertrophy was evaluated using the LV weight (LVW)/body weight (BW) ratio. The myocardial infarct size of rats was determined by 2,3,5-triphenyltetrazolium chloride (TTC) staining. Rats were anesthetized and disinfected, and the thoracic cavity was opened. Then, 1 mL freshly prepared 0.1% TTC solution (Sigma-Aldrich, St Louis, MO, USA) was injected into the postcava using the 1 mL syringe, and the heart beating was maintained for 3–4 min. Later, heart was removed, rinsed in normal saline to remove the residual blood and rapidly placed at −80°C for 10 min. Heart was then sliced into sections which were fixed in 4% paraformaldehyde for 20–30 min to measure the myocardial infarction area using the ImageJ software.
Hematoxylin & eosin staining and Masson staining
Heart was removed and cut into two pieces along the ligation line and vertical to the longitudinal axis of heart, where the part along ligation suture to the tip was maintained and fixed in 4% neutral-buffered formalin, followed by being dehydrated in the graded solutions of ethanol and embedded in paraffin.
Hematoxylin & eosin (HE) staining was performed as follows: Sections, following the regular de-paraffinizing procedure, being stained with hematoxylin for 3 min and eosin for 3 min, and being mounted, were placed under a light microscope (Olympus, Japan) to observe the pathological changes of myocardial tissues and the myocardial infarction of rats. The myocardial tissue damage was evaluated according to a scoring system as described previously. 16 The scoring system is as follows: 0) nil; 1) minimum (focal myocytes damage); 2) mild (small multifocal degeneration with slight degree of inflammatory process); 3) moderate (extensive myofibrillar degeneration and/or diffuse inflammatory process); and 4) severe (necrosis with diffuse inflammatory process).
Masson staining was performed using the Masson kit provided by JianglaiBio (Shanghai, China). Briefly, each paraffin-embedded heart was cut into sections (5 µm thick) and then stained with Masson’s trichrome. Following procedures above, sections were placed under a light microscope (Olympus, Japan) for observation. Collagen fibers were stained into the blue, while cytoplasm, muscle, fibrin and neuroglia into the red. Collagen volume fraction (CVF) was then calculated by using the following formula: CVF = Blue Area / Total Area × 100%. 17 For each section, four fields were selected randomly for photographing.
Quantitative reverse transcriptase polymerase chain reaction
Myocardial tissues collected from rats in each group were homogenized in a tissue homogenizer (Thermo, USA) and prepared for the extraction of total RNA using the TRIzol reagent (Invitrogen, USA). Next, the concentration and purity of RNA were determined by using the Nano Drop 2000 (Thermo, USA)). Then, PCR reaction system was prepared according to the instruction of kit, and the reaction condition was performed with the ABI PRISM 7500 real-time PCR System (ABI) using SYBR Green I kit (TaKaRa, Japan) to amplify the expression of Collagen I (Col-I) and Collagen III (Col-III). The relative expression of target genes was calculated using the formula of 2−ΔΔCt (ΔΔCT = ΔCtexperiment − ΔCtcontrol, ΔCt = Cttarget gene − Ctβ-actin), with the β-actin as the internal reference. The serum Col-I and Col-III levels were also detected by using the corresponding enzyme-linked immunosorbent assay (ELISA) kits (Shanghai Westang Bio-Tech Inc., Ltd).
Transferase-mediated D-UTP-biotin nick end labeling staining
Paraffinized sections were de-paraffinized and hydrated, followed by the addition of 20 μg/mL of protease K for permeabilization of membrane for 20 min at room temperature. Following three washes in phosphate buffer solution (PBS, 7 min/time), the sections were immersed in 0.3% H2O2 for 30 min and then incubated with 0.1% TritonX-100 on ice for 2 min. Thereafter, sections were washed with PBS for three times (7 min/time) and then stained with the TUNEL reaction solution in dark at 37°C for 1 h. The sections were then rinsed twice in Buffer A, blocked in 50 µL bovine serum albumin (BSA) at 37°C for 30 min and incubated with 40 µL color-substrate solution at 37°C for 1 h. In Buffer B, sections were then rinsed and incubated with chromogenic reaction liquid in a dark box for 20 min followed by the addition of 4′, 6-diamidino-2-phenylindole (DAPI) solution for 10 min incubation at 37°C and mounted in 50% glycerinum. Subsequently, sections were placed under a fluorescent microscope in a dark room to observe the changes in apoptosis.
Western blotting
Tissue homogenizer (Thermo) was used to grind the tissue samples and the total protein was extracted, followed by determination of the protein concentrations using the bicinchoninic acid (BCA) kit (Boster Biological Technology Co. Ltd., Wuhan, China). Then, the protein samples were boiled with the loading buffer at 95°C for 10 min and loaded into the wells (40 μg/well), followed by 10% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) (Boster Biological Technology Co. Ltd., Wuhan, China) to isolate the proteins in stacking gel at 80 V and then separation gel at 120 V which were later transferred into the Polyvinylidene Fluoride (PVDF) membrane at 100 mV for 90 to 120 min. Subsequently, the membranes were blocked in 5% BSA at room temperature for 1 h, and then incubated with the primary antibodies including anti-NDRG2 (Santa Cruz), TGF-β1 (ab92486, Abcam), TIMP-2 (ab230511, Abcam), p-Smad2 (ab53100, Abcam), Smad2 (ab63576, Abcam), p-Smad3 (ab52903, Abcam) and Smad3 (ab40854, Abcam) at 4°C overnight. Next, the membranes were incubated with the corresponding secondary antibodies at room temperature for 1 h. At last, membranes were placed into the chemiluminescence (ECL) reagent for the visualization of bands, and the gray value was analyzed for the target bands with Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the loading control by using the Image J software.
Statistical analysis
All data were processed in SPSS 18.0 software (SPSS, Inc, Chicago, IL, USA). Measurement data were expressed in form of mean ± standard deviation (x ± SD), and those in normal distribution among groups by one-way analysis of variance (ANOVA), followed by Tukey’s post hoc test for multiple comparisons. p < 0.05 suggested that the difference had statistical significance.
Results
Echocardiography and hemodynamic parameters of rats in each group
As shown in Figure 1(a) and (b), rats manifested the following changes in echocardiography. In comparison with the Sham group, rats in the Model group had sharp decreases in LVEF and LVFS, while when compared to Model group, rats in the Model + AdNDRG2 group had obvious increases in LVEF and LVFS (all p < 0.05). As indicated by hemodynamic parameters (Figure 1(c) to (f)), rats in the Model group had a higher LVEDP but lower LVSP and ±dp/dt max than those in the Sham group (all p < 0.05). The comparison between the Model group and Model + AdEGFP group showed no significant difference in the levels of the indicators above (all p > 0.05). Nevertheless, rats in the Model + AdNDRG2 group manifested the obvious decrease in LVEDP, but significant increases in LVSP and ±dp/dt max (all p < 0.05).

Comparison of the echocardiography and hemodynamic parameters of rats among groups. (a) Comparison of LVEF and (b) LVFS of rats measured by echocardiography. (c) Original record showing the left ventricular pressure tracing in one Sham rat. (d) Comparison of ±dp/dt max, (e) LVEDP, and (f) LVSP of rats obtained by hemodynamic evaluation;
Comparison of the myocardial infarction area of rats among groups
No significant differences in body weight were found among the four groups at the end of the study (Figure 2(a)). However, the LVW/BW ratio was higher in the Model group compared with Sham group (p < 0.05). While the Model + AdNDRG2 group showed lower LVW/BW ratio as compared with the Model group (p < 0.05, Figure 2(b)). TTC staining showed that normal myocardial tissues were in brick-red, while the infarcted tissues were in white (Figure 2(c)). As compared with the Sham group, rats in the Model group had a significant enlargement in the infarct size (p < 0.05). However, rats in the Model + AdNDRG2 group had a sharp decrease concerning the infarct size (p < 0.05), but no significant alterations were found in the Model + AdEGFP group (p > 0.05, Figure 2(d)).
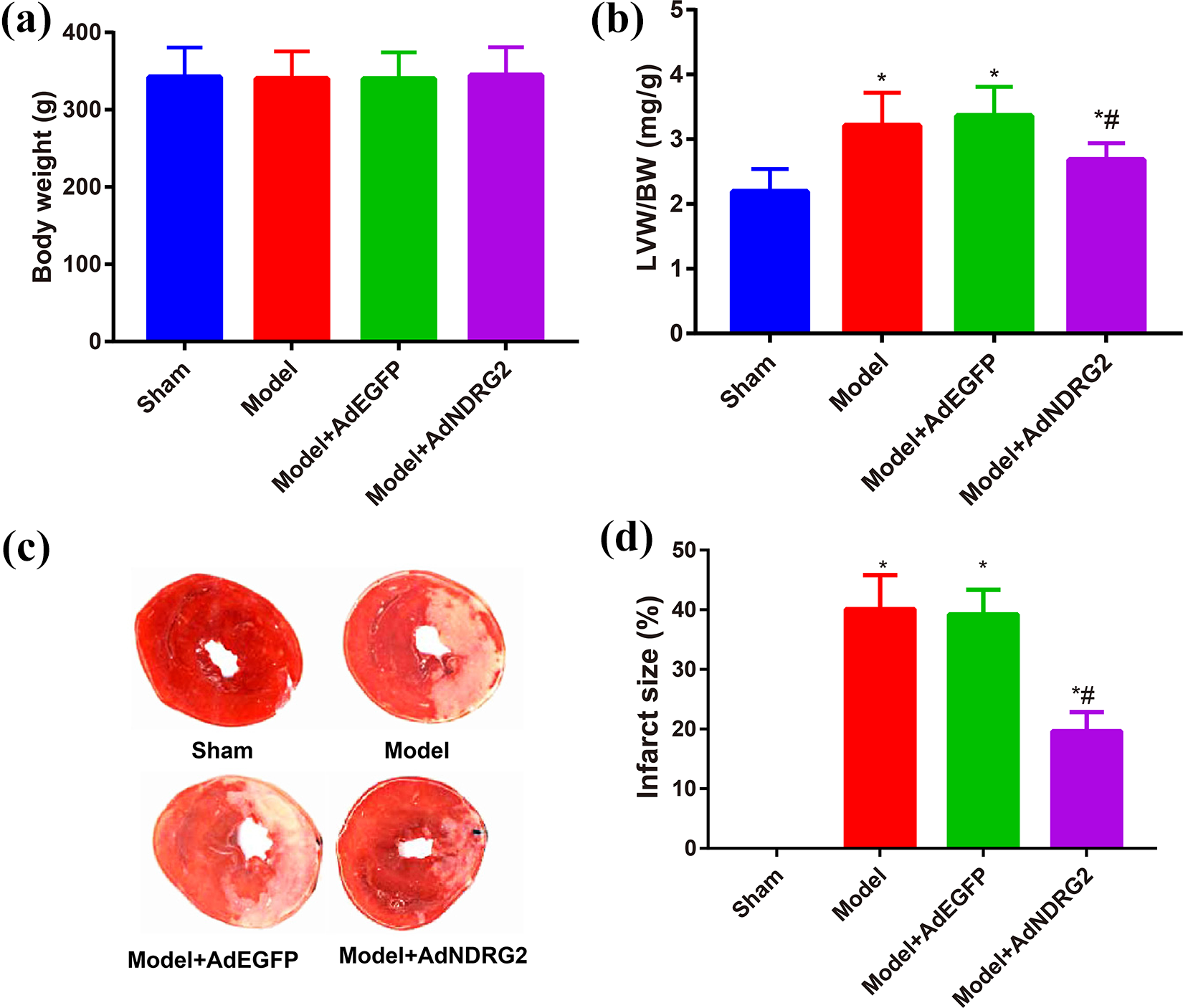
Comparison of the LVW/BW ratio and myocardial infarction area of rats among groups. (a) Comparison of the body weight and (b) LVW/BW ratio of rats among groups. (c) The myocardial infarct size of rats among groups was determined by TTC staining;
Observation of HE and Masson staining of rats among groups
HE staining and Masson staining (Figure 3(a)) showed that rats in the Sham group had well aligned myocardial fibers, with no evident collagen deposition or infiltration of inflammatory cells, while those in the Model group showed a disordered arrangement of myocardial fibers, with massive collagen deposition and infiltration of inflammatory cells. Rats in the Model + AdNDRG2 group had poorly aligned, hypertrophic myocardial fibers, with amelioration in the collagen deposition and infiltration of inflammatory cells. Furthermore, in comparison with the Sham group, rats in the Model group had an obvious increase in the damage score and CVF (all p < 0.05). When compared with the Model group, the damage score and CVF of rats in the Model + AdNDRG2 group decreased magnificently (p < 0.05, Figure 3(b) and (c)).

Observation of HE staining and Masson staining of rats among groups. (a) Representative images of HE and Masson staining of rats. (b) Comparison of the damage score and (c) CVF of rats among groups
Expression of Col-I and Col-III in myocardial tissues of rats
According to the results of qRT-PCR and ELISA shown in Figure 4, the expression of Col-I and Col-III in myocardial tissues of rats in the Model group was up-regulated in comparison with the Sham group (all p < 0.05), but no significant change was detected in the expression of Col-I and Col-III in myocardial tissues of rats from the Model + AdEGFP group, as compared to the Model group, (all p > 0.05), and on the contrary, rats in the Model + AdNDRG2 group had sharp decreased levels of Col-I and Col-III (all p < 0.05).

Comparison of the expression of Col-I and Col-III in myocardial tissues of rats among groups. (a) Expression of Col-I and Col-IIII in the myocardial tissues of rats in each group detected by qRT-PCR and (b) ELISA.
Apoptosis of myocardial tissues of rats in each group
TUNEL staining was carried out to determine the apoptosis of myocardial tissues (Figure 5), and the results indicated that in comparison with the Sham group, rats in the Model group had a sharp increase in the apoptotic rate of myocardial tissues (all p < 0.05), while as compared to the Model group, rats in the Model + AdEGFP group had no significant difference in the apoptotic rate (all p > 0.05), and at the same time, those in the Model + AdNDRG2 group experienced a sharp decrease (all p < 0.05).

Apoptosis of myocardial tissues of rats in each group determined by TUNEL staining.
Expression of NDRG2 and TGF-β1/Smad pathway of rats in each group
Western blotting was applied to detect the protein expression of NDRG2 and TGF-β1/Smad pathway. As indicated in Figure 6, NDRG2 was down-regulated in the Model group, while the expression of TGF-β1, p-Smad2/Smad2, p-Smad3/Smad3 and TIMP-2 was up-regulated as compared to the Sham group (all p < 0.05). However, the treatment of AdNDRG2 for rat models could reverse the changes above (all p < 0.05).

Expression of NDRG2 and TGF-β1/Smad pathway of rats in each group detected by Western blotting assay.
Discussion
It has been reported that any severe and continuous changes in the hemodynamics of heart could result in the pathophysiological outcome, like myocardial remodeling and damage to the myocardial cells. 18,19 In general, the hemodynamic parameters (like ±dp/dtmax, LVEDP and LVSP) and echocardiography (including LVEF and LVFS) have been confirmed to precisely evaluate the changes in the heart structure and function of rat models. 20 As documented, the decline in myocardial systolic function comes with decreases in LVEF, LVFS and ±dp/dtmax, as well as the reduced LVSP and the elevated LVEDP. 21 Similar results were also found in the CHF models of the current study, demonstrating a severe impairment in cardiac function, 22 which characterizes the development of CHF, indicating the successful establishment of our rat models. Importantly, the overexpression of NDRG2 could ameliorate the indicators above. Consistently according to the work of Tang et al., the levels of LVEF, LVFS, ±dp/dtmax and LVSP were increased with the declined LVEDP via overexpression of SDF-1, thereby improving cardiac structure and function, and reducing the myocardial infarction area of rats, 23 also suggesting a beneficial effect of NDRG2 on cardiac function in HF rats.
Previous evidence has confirmed that the apoptosis of myocardial cells is a critical element in HF, with remarkable decrease in the quantity of myocardial cells and progressive exacerbation in fibrosis, thereby leading to the myocardial remodeling and deteriorating the heart function. 24 Studies have also shown that heart failure rats present with an evident increase in the apoptosis of myocardial cells. 25 Our findings supported that the myocardial tissues of rat models manifested significantly increased apoptosis and enhanced infarct size, which, however, was reversed by the overexpression of NDRG2. Recent studies have unveiled crucial roles of NDRG2 in many physiological and pathological conditions, such as cell proliferation, differentiation, and stress response. 26,27 In agreement, possibly by activating NDRG2, the pathological changes and release of inflammatory cytokines in the lung tissues after the attack of intestinal ischemia-reperfusion injury were improved with the pre-treatment of Melatonin (MT). 28
It was also interesting to note that, NDRG2 gene deletion could result in the increase of cerebral infarct volume in rats with I/R injury, indirectly confirming the neuroprotective role of NDRG2 in cerebral I/R injury. 29 Also, in the study of Sun et al., the development of myocardial ischemia/reperfusion injury was exhibited with the increased apoptosis of myocardial cells and the down-regulation of NDRG2, 30 and knockout of NDRG2 can increase the apoptosis of myocardial cells and the infarction area during this period, 10 indicating that overexpression of NDRG2 could protect the myocardial cells from injury in HF rats.
As indicated, apoptosis or necrosis in myocardial tissues due to the pathological changes would deprive the ability of regeneration from myocardial cells, and, thus, tissues in lesion would be filled by the extracellular matrix, resulting in the increased generation of collagen and alteration of the heart structure and function. 31,32 Col-I and Col-III are the two most common types of collagens in the extracellular matrix of myocardial cells. 33 In this work, we found that rats in the Model group had significant up-regulation of Col-I and Col-III and an increase in the CVF of myocardial tissue, while overexpression of NDRG2 abolished the changes above, suggesting that NDRG2 could improve the abnormal collagen deposition in the extracellular matrix of myocardial cells and inhibit the myocardial fibrosis in CHF rats, thus ameliorating the myocardial remodeling. Jin et al. reported that knockout of NDRG2 would reverse the increased Col-I and Col-III induced by TGF-β1 to promote the renal fibrosis, while overexpression of NDRG2 ameliorated the renal fibrosis. 34 Furthermore, overexpression of NDRG2 in our study was able to down-regulate the expression of TGF-β1, p-Smad2/Smad2, p-Smad3/Smad3 and TIMP-2. TGF-β1 is a locally generated cytokine that has been implicated as a major stimulator of tissue fibroinflammatory changes and plays an important role in the pathogenesis of cardiac remodeling and fibrosis. 35,36 Physiologically, TGF-β1 binds to the latency associated peptide (LAP) to form the inactive complex that fails to bind to the receptor, 37 while the injury of heart would transform the inactive TGF-β1 outside the cells rapidly into the active form. 38 Smads family represents the key intracellular effectors of TGF-β1, and the ligand of TGF-β1 could bind to Type II/Type I receptor to facilitate the phosphorylation of Smad2/3 which further binds to Smad4; and at the meantime, the resulting complex is translocated into the nucleus to up-regulate the expression of extracellular matrix, including α-SMA, collagen I and collagen III, eventually causing the tissue fibrosis or remodeling. 39 –41 Furthermore, TGF-β stimulation induces myofibroblast differentiation and enhances extracellular matrixprotein synthesis by inhibiting MMP expression and inducing synthesis of protease inhibitors, such as TIMP. 42,43 Consistently, He et al. found the TGF-β/Smad pathway inhibited by HMGB1 could suppress the expression of Col-I, Col-III and TIMP-2, thereby limiting the ventricular remodeling and improving the myocardial fibrosis. 44 Findings of Yang et al. also uncovered that overexpression of NDRG2, by inhibiting the activity of TGF-β1/Smad, could reduce the expression of TIMP2, thereby improving the liver function of rats with liver fibrosis. 14 Therefore, overexpression of NDRG2 may inhibit the TGF-β1/Smad pathway to suppress the myocardial fibrosis, reduce the collagen formation and improve the myocardial remodeling of HF rats.
In summary, NDRG2 could reduce the apoptosis of myocardial cells and improve the cardiac function and myocardial remodeling in HF rats via inhibiting the activity of TGF-β1/Smad pathway. Thus, it is conceivable that modulating NDRG2 expression may provide a novel therapy for intervention in HF.
Footnotes
Acknowledgements
The authors would like to thank all the reviewers in this paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.