
Abstract
Propofol is a commonly used drug for sedation and general anesthesia during cancer surgery. Previous studies indicate that propofol exerts anti-tumor effect in various cancers. The aim of this study was to investigate the underlying molecular mechanism of propofol in liver cancer. The effects of propofol on liver cancer cells were evaluated by cell viability assay, colony formation assay, and tumor xenograft model. Dysregulated lncRNAs of propofol-treated liver cancer cells were evaluated by transcriptome RNA sequencing. The underlying molecular mechanisms of lncRNA cancer susceptibility candidate 9 (CASC9) in propofol-induced anti-tumor effects were evaluated by western blot, quantitative real-time polymerase chain reaction (qRT-PCR), wound scratch healing assay, transwell cell migration and invasion assay, TUNEL staining, fluorescence in situ hybridization, RNA immunoprecipitation (RIP), and chromatin immunoprecipitation (ChIP). We found that propofol suppressed proliferation, migration, invasion, and tumor xenograft growth of liver cancer cells in a dose-dependent manner. Exosomes transfer from propofol-treated cells inhibited proliferation, migration, and invasion and promoted apoptosis of liver cancer cells. Transcriptional profiling of propofol-treated liver cancer cells identified CASC9 as significantly downregulated lncRNA in cells and exosomes. Enforced CASC9 expression partially rescued the inhibitory effects of propofol on liver cancer cells. Furthermore, CASC9 was found to interact directly with EZH2 and epigenetically regulated PTEN expression. Restoration of CASC9 partially abrogated the inhibition of propofol on Akt/mTOR signaling. Our results indicated that propofol exerted anti-tumor effects by downregulating CASC9, and subsequently suppressed Akt/mTOR signaling. Our findings provided a novel insight into propofol-induced anti-tumor effects in liver cancer.
Introduction
Liver cancer is the second leading cause of cancer-related death worldwide. Primary liver cancer can be divided into hepatocellular carcinoma, intrahepatic cholangiocarcinoma, fibrolamellar carcinoma, hepatoblastoma, or other rare cancers. 1 Among them, hepatocellular carcinoma (HCC) accounts for more than 80% of all liver cancer cases; thus, liver cancer is mainly referred to HCC in our study. The major risk factors for liver cancer are chronic hepatitis B or C infection, alcohol consumption, nonalcoholic fatty liver disease, and contamination of dietary aflatoxins and aristolochic acid. 2 Due to late diagnosis and recurrence, the prognosis of liver cancer is really poor, with a 5-year survival rate as low as 18%. 3 Virtually, more than 70% liver cancer patients will eventually experience cancer recurrence in 5 years’ time after curative treatment. 2 Liver cancer is known resisting to nearly all conventional chemotherapeutic drugs, especially at advanced stages. Thus, it is important to explore the underlying molecular mechanisms of tumorigenesis of liver cancer and find new agents for liver cancer treatment.
Propofol (2,6-diisopropylphenol) is an intravenous hypnotic drug frequently used for sedation and implementation of general anesthesia during cancer surgery. Retrospective studies reveal a protective role of propofol in cancer patients after surgery. For example, a retrospective study of 2856 gastric cancer patients suggests that propofol-based total intravenous anesthesia may improve survival of gastric patients compared with sevoflurane-based inhalational anesthesia. 4 In colon cancer, propofol anesthesia shows a more favorable survival compared with desflurane anesthesia for colon cancer surgery. 5 Although these studies have a small number of enrolled patients and may subject to bias, they raise a possibility of potential benefits for propofol-based anesthesia in cancer surgery. A number of preclinical studies have proved the anti-tumor effects of propofol in various cancers, such as that in breast, colon, cervical, and lung. 6 Propofol also shows potential anti-tumor effect in liver cancer. For example, a retrospective study suggests that propofol-based anesthesia is correlated with improved survival of HCC patients who underwent hepatectomy. 7 Besides, a number of studies in HCC cell lines or animal models demonstrate the inhibitory effects of propofol on proliferation, migration, and invasion of HCC cells.8–11 Albeit several possible mechanisms for the anti-tumor effects of propofol in liver cancer have been raised, these mechanisms might not fully elucidate the function of propofol in liver cancer. Thus, we conducted transcriptome RNA sequencing analysis to search for possible long non-coding RNAs (lncRNAs) that were involved in the anti-tumor effects of propofol in liver cancer.
LncRNAs are a wide-spread category of RNA transcripts longer than 200 nucleotides but without protein-coding potential. Although the expression levels of lncRNAs are lower than protein-coding genes, they are more tissue specific and exhibit various cellular functions. A number of studies prove that lncRNAs play a role in the anti-tumor effects of propofol. For example, propofol treatment reduces HOTAIR expression in colon cancer cells, while forced HOTAIR expression abrogates propofol-induced apoptosis and inhibition on cell invasion. 12 In HCC, propofol treatment increases the expression level of lncRNA DGCR5, thus restrains proliferation, migration, and invasion of HCC cells via inactivating Raf1/ERK1/2 and Wnt/β-catenin pathways. 13 Exosomes are nano-sized microvesicles that transport signals or materials from cell to cell. These materials include nucleic acids, proteins, and lipids. Notably, lncRNAs can be carried by exosomes and dispersed from cell to cell, thus exert various functions. For instance, propofol reduces the level of lncRNA H19 in exosomes and, thus, suppresses proliferation and metastasis of HCC cells. 14 Exosomes are proved to participate in establishing the micro environment of liver cancer in recent years; 15 thus, we also evaluated the influence of propofol on exosomes in the present study.
Cancer susceptibility candidate 9 (CASC9) is a newly found lncRNA that locates at 8q21.11. CASC9 is dysregulated and involved in the development of multiple cancers, including liver cancer.16–19 In our study, transcriptional profiling of propofol-treated liver cancer cells identified CASC9 as significantly downregulated lncRNA. Further study revealed that CASC9 overexpression rescued the inhibitory effects of propofol on proliferation, migration, invasion, and tumor growth by epigenetically suppressing phosphatase and tensin homolog (PTEN) and subsequently activating Akt/mTOR pathway. Our results elucidated the underlying molecular mechanisms of propofol-induced anti-tumor effects in liver cancer.
Materials and methods
Patient samples
The experiments involved human subjects were reviewed and approved by the Ethics Committee of Heilongjiang Provincial Hospital (SYXK(Hei)2015-013). Informed consents were obtained from all patients participated in our study. A hundred pairs of liver cancer tissues and adjacent normal tissues were collected from Heilongjiang Provincial Hospital between May 2015 and June 2016. Patients’ data were retrieved from the hospital database, and follow up was continued for as long as 48 months post-surgery.
Cell culture and reagents
Liver cancer cell lines SNU-449 and HepG2, and human HEK293 T cells were obtained from American Type Culture Collection (ATCC). Liver cancer cell lines HuH-6 and HuH-7 were obtained from Japan Health Science Research Resources Bank. All cell lines were cultured with RPMI-1640 (Invitrogen, USA), supplemented with 10% fetal bovine serum (Hyclone, USA), and 1% penicillin/streptomycin (Gibco, USA) in a humidified atmosphere at 37°C containing 5% CO2. Propofol (Sigma #BP1031, USA) was dissolved in DMSO (50 mg/mL) and diluted in culture medium to achieve the final concentrations of 10, 25, and 50 μg/mL. DMSO was used as vehicle control.
Plasmid constructs, lentivrius package, and infection
The full length of CASC9 was cloned into the pCDH lentivrius vector (System Biosciences #CD510B-1). The empty pCDH vector was used as empty vector (EV) control. Short hairpin RNAs (shRNA) targeting CASC9 (sh-CASC9-1 and sh-CASC9-2) were inserted into the pLKO.1 plasmid. The pLKO.1 plasmid inserted with a non-targeting sequence was used as sh-NC control. Short guide RNAs (sgRNA) targeting EZH2 (sgEZH2-1 and sgEZH2-2) were inserted into the lentiCRISPRv2 vector (Addgene #52961). The empty lentiCRISPRv2 vector inserted with a non-targeting sequence was used as sg-NC control. The sequences for shRNAs and sgRNAs were as follows: sh-CASC9-1, 5′-TTGAG AAGTT AGAAT GTGAT TTT-3′, sh-CASC9-2, 5′-GGCAA GAAAC AGGTT ATGTT TGG-3′, sgEZH2-1, 5′-CATCA TCATT ATATT GACCA-3′, sgEZH2-2, and 5′-TTTAC ATAAC ATTCC TTATA-3’. Lentivirus package was conducted by introducing lentivirus plasmids with helper plasmids into HEK293 T cells using lipofectamine 3000 (Invitrogen, USA). Virus-containing medium was harvested at 24, 48, and 72 h post-transfection, then stored at −80°C for further use. Lentivirus infection was carried out by incubating cells with virus-containing medium overnight with 8 μg/mL polybrene (Sigma, USA).
Cell viability assay
Viability of cells was assayed as described previously using the CellTiter-Glo kit (Promega #G7572). 20 Cells (5000/well) were seeded in 96-well plates and treated as indicated. Then cells and CellTiter-Glo reagents were equilibrated to room temperature, mixed thoroughly on an orbital shaker for 5 min, and placed in a dark place for 15 min to stable the luminescence signal. Next, recorded the luminescence signal on a microplate reader. All samples were repeated in triplicate.
Colony formation assay
Colony formation assay was performed as described previously. 20 SNU-449 and HuH-6 cells transducing with CASC9 or EV control were treated with propofol (25 μg/mL) or equal volume of DMSO (Veh) for colony formation assay. Cells were dispersed as single cell suspensions, then seeded in 6-well plates (2000/well) for 3 weeks without disturbance. Colonies were fixed with 4% paraformaldehyde for 15 min, stained with crystal violet for 1 h at room temperature, and then viewed and photographed by a microscope. All samples were repeated in triplicate.
Western blot
The protocol for western blot was conducted as described previously. 20 Culture cells were digested by RIPA buffer containing protease inhibitors (Sigma, USA). Cell lysates were collected by centrifuge. Protein concentration was measured by BCA kit (Thermofisher, USA) and separated by 10% SDS-PAGE, then transferred to nitrocellulose membranes. Next, the membranes were blocked by 5% non-fat milk for 1 h, then incubated with specific first antibodies at 4°C overnight and corresponding second antibodies for 1 h at room temperature. The antibodies used in our study were PCNA (Cell signaling #13110, 1: 1000), GAPDH # (Cell signaling 5174, 1: 1000), CD9 (Cell signaling #13403, 1: 1000), CD81 (Cell signaling #56039, 1: 1000), PTEN (Cell signaling #9188, 1: 1000), EZH2 (Cell signaling #5246, 1: 1000), p-Akt (Ser473) (Cell signaling #4060, 1: 1000), Akt (Cell signaling #9272, 1: 1000), p-mTOR (Ser2448) (Cell signaling #5536, 1: 1000), mTOR (Cell signaling #2972, 1: 1000), and Goat Anti-Rat IgG (HRP Conjugate) (Cell signaling #5536, 1: 4000).
Tumor xenograft model
The protocols for animal studies were reviewed and approved by the ethics committee of Heilongjiang Provincial Hospital. The dosage of propofol was referred to previous study. 21 Parental SNU-449 cells or SNU-449 cells transduced with EV or CASC9 (2×106) were subcutaneously injected into nude mice, and then tumor xenografts were allowed to grow for 2 weeks. Mice were randomly divided into Vehicle group (Veh) and propofol treatment group (10, 25, or 50 mg/kg) (n = 4 for each group), or Veh + EV, Veh + CASC9, and Pro + CASC9 group (25 mg/kg) (n = 5 for each group). Propofol treatment group (10, 25, or 50 mg/kg) received 10, 25, or 50 mg/kg propofol treatment. Pro + CASC9 received 25 mg/kg propofol treatment, while Veh + EV and Veh + CASC9 group received an equal volume of soybean oil. Propofol was dissolved in soybean oil and injected intraperitoneally into nude mice daily for 3 weeks. Tumor growth was measured every 3 days and calculated by the formula (length × width 2 )/2. At the end of drug treatment, mice were sacrificed and tumors were dissected out and weighed.
Transcriptome RNA sequencing and data analysis
Transcriptome RNA sequencing was carried out as described previously. 22 SNU-449 and HuH-6 cells (5 × 105) were seeded in 6-well plates and treated with 25 μg/mL propofol or equal volume of DMSO for 48 h, then cells were collected for total RNAs extraction using RNeasy Mini kit (Qiagen). NEB Next Ultra II Directional RNA Library Prep kit (New England BioLabs) was used to generate RNA-seq libraries. The libraries were sequenced by NextSeq 500. Raw data for gene expression were quantified by Salmon v0.8.2. DESeq2 was used for data analysis. Two-way ANOVA was used to evaluate significant different expressed genes. p value <0.05 and |log2 Fold Change| ≥ 2 were used to select dysregulated lncRNAs.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNAs were extracted by TRIzol reagent (Invitrogen, USA) as protocol indicated. RNA concentration was evaluated by NANO drop 2000. PrimeScript RT Reagent Kit (TakaRa, China) was used to synthesize the complementary DNA. qRT-PCR was performed by SYBR Premix Ex Taq™ II kit (TakaRa, China). Data were analyzed using the 2–ΔΔCt method. 23 GAPDH was used as internal control. The primer sequences for qRT-PCR were listed below. CASC9, forward, 5′-GGCTG GAGAG TCATT GGCAC TA-3′, reverse, 5′-TCCAT TGCTT TGCTG CTGTC TG-3’; GAPDH, forward, 5′-TGGCA CCGTC AAGGC TGAGAA-3′, reverse, 5′-TGGTG AAGAC GCCAG TGGAC TC-3’. All samples were repeated in triplicate.
Exosome isolation and identification
Exosomes were isolated as described previously. 24 SNU-449 or HuH-6 cells were treated with 25 μg/mL propofol, then medium in 10 cm culture dishes were harvested and centrifuged at 3000 g for 15 min, then filtered by 0.22 mm filter (Millipore, USA). Exosomes were extracted using the Exoquick exosome precipitation solution (System Biosciences, China). After incubating with the exosome precipitation solution for 24 h, exosomes were obtained by centrifuging at 1500 g for 30 min and then resuspended in phosphate buffered saline (PBS) solution. Successful extraction of exosomes was verified by western blot analysis of two exosome markers CD9 and CD81.
Wound scratch healing assay
SNU-449 and HuH-6 cells transducing with CASC9 or EV control were treated with propofol (25 μg/mL) or equal volume of DMSO (Veh) for wound scratch healing assay. Cells (5 × 105) were seeded in 6-well plates. A scratch was made by a sterile plastic tip right in the middle of the well when cell confluence reached 70%. Washed out cell debris by PBS, then cells were viewed and photographed by microscope at 0 and 24 h. Digimizer software system (MedCalc software, Belgium) was used to calculate the migration distance.
Transwell cell migration assay
To evaluate cell migration, cells (5 × 104/well) were seeded in the upper chamber (Costar Corp, USA) with 0.5% fetal bovine serum. The lower chamber was filled with culture medium containing 20% fetal bovine serum. Cells were allowed to migrate for 36 h. To evaluate cell invasion, cells (1 × 105/well) were used, and the filter was pre-coated with matrigel (BD Biosciences, USA). The other procedures were same as migration assay. The migration or invasion cells were stained by crystal violet.
TUNEL staining
One step TUNEL apoptosis assay kit (Beyotime #C1090, China) was used to detect apoptotic cells as protocol indicated. Cells (5×105) were seeded in 6-well plates, fixed by 4% paraformaldehyde for 15 min and permeabilized by 0.25% Triton-X 100 for 30 min at room temperature. Cells were then incubated with terminal dexynucleotidyl transferase (TdT) reaction cocktail for 60 min at 37°C avoiding light, washed with PBS for three times, and photographed by LSM 5 Pa Laser Scanning Microscope (Zeiss Germany, Oberkochen, Germany). DAPI was used to stain the nucleus.
Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed as described previously.23,25 Cells (1 × 104) were seeded on coverslips for 48 h, then fixed by 4% paraformaldehyde for 15 min, and permeabilized by 0.5% Triton X-100 for 30 min. CASC9 probe was synthesized by mMESSAGE T7 Ultra In Vitro Transcription kit (Life Technologies, USA) and labeled with FITC-UTP (Roche, Switzerland). Cells were incubated with CASC9 probe for 16 h at 37°C, then washed with 50% formamide/2 × saline sodium citrate. DAPI was used to stain the nucleus. Images were obtained under a confocal laser-scanning microscope (LSM 780, Zeiss).
RNA immunoprecipitation
Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA) was employed for RIP assay as described previously. 26 In brief, cell lysates from 2 × 107 cells were incubated with beads for 8 h, and washed with RIP wash buffer. The EZH2 antibody (Cell signaling #5246, 1: 50) and IgG (Cell signaling #3900, 1: 50) were used for RIP assay. The immunoprecipitated RNA was evaluated by qRT-PCR.
Chromatin immunoprecipitation
EZ-ChIP kit (Millipore, USA) was used for ChIP assay as described previously. 27 Cells (1 × 107) were treated with formaldehyde for 10 min and then sonicated to DNA fragments. Next, cells were incubated with EZH2 antibody (Cell signaling #5246, 1: 50) or H3K27me3 antibody (Cell signaling #9733, 1: 50). The precipitated DNA was evaluated by qRT-PCR.
Statistical analysis
Statistical analysis was performed using SPSS 16.0 or GraphPad Prism 8.0. The difference between groups was evaluated by Student’s t test or one-way ANOVA (Tukey’s post-hoc test). Kaplan–Meier method was used to evaluate overall survival with log-rank test. Half maximal inhibitory concentration (IC50) was calculated using the nonlinear regression model of GraphPad Prism 8.0. p < 0.05 was considered statistically significant.
Results
Dose-dependent inhibition of proliferation, migration, invasion, and tumor growth of liver cancer cells by propofol
To evaluate the cytotoxicity of propofol, two commonly used liver cancer cell lines SNU-449 and HuH-6 were exposed to a series concentration of propofol for cell viability assay. As depicted in Figure 1(a), propofol reduced cell viability of SNU-449 and HuH-6 cells in a dose-dependent manner. The IC50 of propofol was 38.5 μg/mL for SNU-449 and 26.7 μg/mL for HuH-6, respectively. According to the IC50 of propofol, low, middle, and high concentrations of propofol (10, 25, and 50 μg/mL) were chose. The influence of propofol on proliferation of liver cancer cells was evaluated by cell viability assay, colony formation assay, and western blot. We found that propofol suppressed growth of SNU-449 and HuH-6 cells in a time- and dose-dependent manner (Figure 1(b)). Besides, propofol dose-dependently inhibited the clonogenic growth of SNU-449 and HuH-6 cells in colony formation assay (Figure 1(c) and (d)). In addition, propofol suppressed migration and invasion of SNU-449 and HuH-6 cells (Figure 1(e) and (f)). PCNA is a marker of cell proliferation. In western blot analysis, propofol reduced the expression of PCNA (Figure 1(g)). The effect of propofol was also evaluated in nude mice in vivo. We found that propofol restrained tumor xenograft growth of SNU-449 cells, with decreased tumor volume and weight (Figure 1(h)–(j)). Taken together, our results indicated that propofol suppressed proliferation, migration, invasion, and tumor xenograft growth of liver cancer cells in a dose-dependent manner.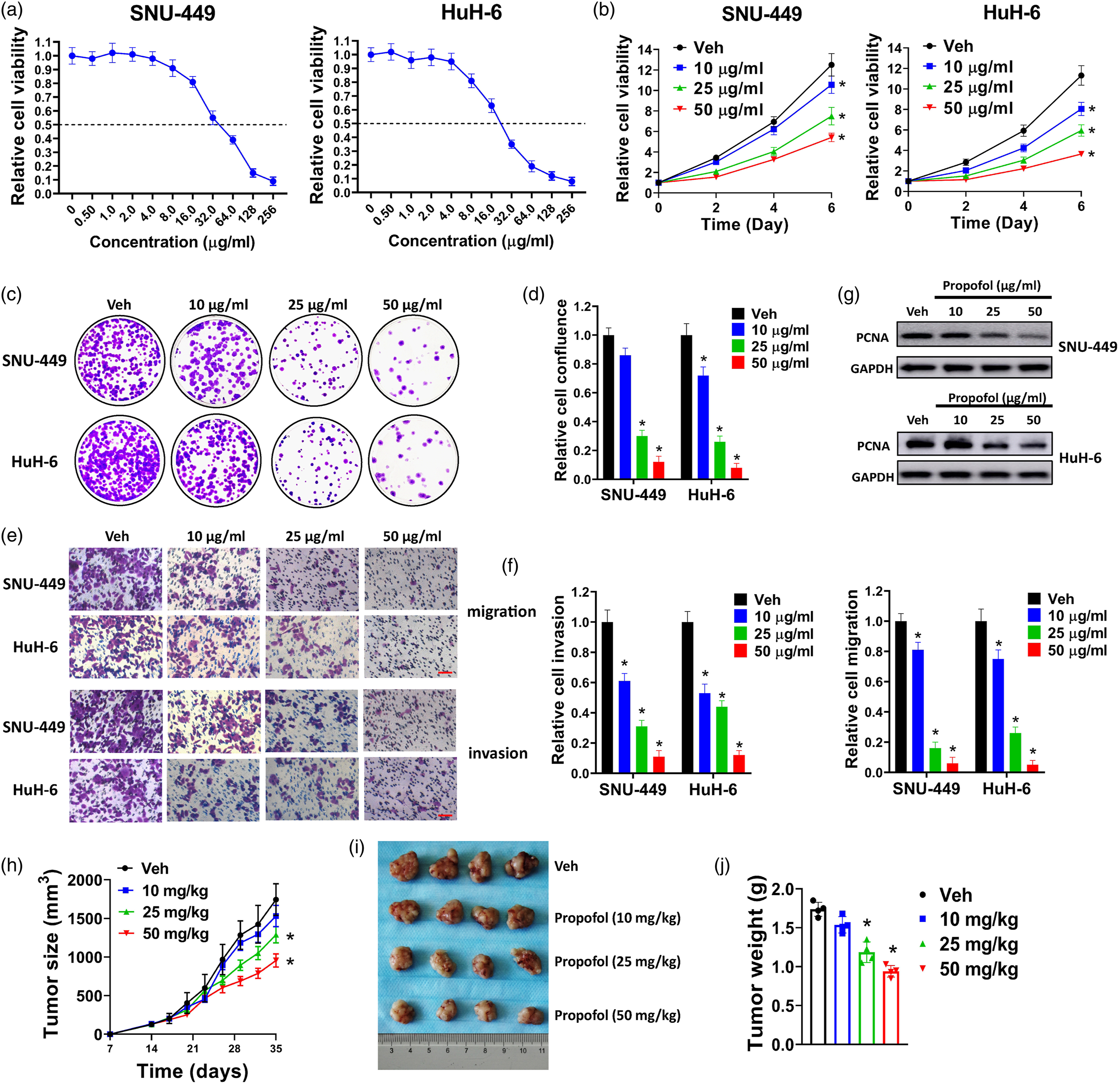
Exosomes from propofol-treated cells inhibit proliferation, migration, and invasion and promote apoptosis of liver cancer cells
Exosomes from propofol-treated SNU-449 and HuH-6 cells were extracted. CD9 and CD81 are markers for exosomes. Compared with parental cell control, exosomes showed high levels of CD9 and CD81 (Figure 2(a)). Next, two commonly used liver cancer cell lines HepG2 and HuH-7 were incubated with exosomes from propofol-treated SNU-449 cells (Pro-exo). Exosomes from vehicle-treated (DMSO) SNU-449 cells were used as control (Veh-exo). HepG2 and HuH-7 cells incubated with Pro-exo showed reduced cell growth compared with cells incubated with Veh-exo (Figure 2(b)). In wound scratch assay and transwell cell invasion assay, HepG2 and HuH-7 cells incubated with Pro-exo exhibited decreased migration and invasion (Figures 2(c)–(f)). Furthermore, Pro-exo treatment increased the number of TUNEL positive cells in HepG2 and HuH-7 cells (Figure 2(g) and (h)). Taken together, our results indicated that exosomes from propofol-treated cells inhibited proliferation, migration, and invasion and promoted apoptosis of liver cancer cells.
Transcriptome RNA-sequencing of propofol-treated liver cancer cells identifies lncRNA CASC9 as significantly downregulated lncRNAs
To explore potential lncRNAs that were involved in the inhibitory effects of propofol in liver cancer, SNU-449 and HuH-6 cells were treated with propofol and total RNAs were collected for transcriptional RNA sequencing. A total of 557 lncRNAs were found to upregulate or downregulate in SNU-449 cells (Supplementary Table 1), and 791 lncRNAs in HuH-6 cells (Supplementary Table 2), respectively. Among them, 77 lncRNAs were found to dysregulate in both SNU-449 and HuH-6 cells (Supplementary Table 3). The top 20 upregulated or downregulated lncRNAs in SNU-449 and HuH-6 cells were shown in Figure 3(a). Among them, lncRNA DGCR5 and H19 have been reported to be involved in the anti-tumor effects of propofol by previous studies,13, 14 indicating that our strategy was feasible for searching lncRNAs that were associated with propofol-induced anti-tumor effects. The top 20 upregulated or downregulated lncRNAs were further evaluated by us and we ultimately come to CASC9. This newly found lncRNA exhibited oncogenic functions in various cancers, including hepatocellular carcinoma.
18
Thus, we speculated that downregulation of CASC9 might contribute to propofol-induced proliferation inhibition. At first, we proved that propofol suppressed CASC9 expression in SNU-449 and HuH-6 cells dose-dependently (Figure 3(b)). Next, we found that CASC9 was downregulated in exosomes of propofol-treated SNU-449 and HuH-6 cells in a dose-dependent manner (Figure 3(c)). The expression of CASC9 in 100 pairs of liver cancer tissues and adjacent normal tissues were evaluated by qRT-PCR. Our results indicated that CASC9 was upregulated in liver cancer patients (Figure 3(d)). Retrospective analysis of these patients revealed that high CASC9 levels were associated with poor overall survival (Figure 3(e)). Collectively, our results indicated that lncRNA CASC9 was downregulated by propofol in liver cancer cells and exosomes.
Forced expression of CASC9 partially rescues the inhibitory effects of propofol on liver cancer cells
As CASC9 was downregulated by propofol, we speculated that CASC9 might take part in propofol-induced proliferation inhibition. Forced expression of CASC9 was realized by introducing cells with CASC9 expression lentivirus. Compared with empty vector (EV) control, CASC9 was significantly overexpressed in both SNU-449 and HuH-6 cells (Figure 4(a)). Propofol treatment dramatically reduced the levels of CASC9 in SNU-449 and HuH-6 cells, but this was abrogated by CASC9 overexpression (Figure 4(b)). Next, we found that forced expression of CASC9 partially abolished the inhibitory effects of propofol on growth, colony formation, migration, and invasion of SNU-449 and HuH-6 cells (Figures 4(c)–(g)). In western blot analysis, propofol treatment decreased the level of PCNA, but this was replenished by overexpression of CASC9 (Figure 4(h)). In tumor xenograft model, forced expression of CASC9 partially abolished the suppression of propofol on tumor growth, volume, and weight of SNU-449 cells (Figure 4(i)–(k)). Above all, our results indicated that forced expression of CASC9 partially rescued the inhibitory effects of propofol on liver cancer cells.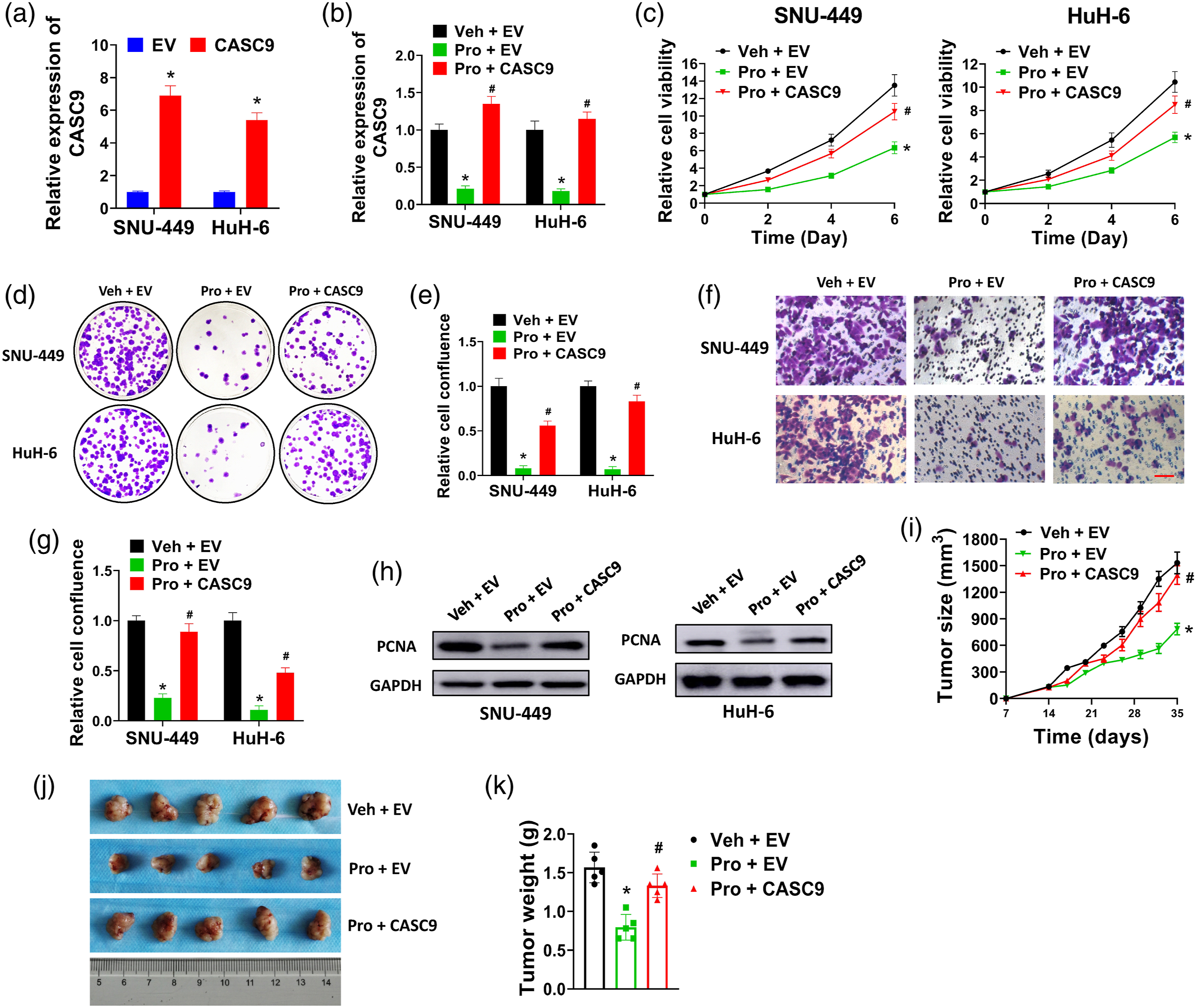
CASC9 epigenetically mediates PTEN expression by interacting with EZH2
As CASC9 played an important role in propofol-induced proliferation inhibition, the underlying molecular mechanism of CASC9 in liver cancer had been explored. Subcellular location is vital for lncRNAs to exert their functions. In qRT-PCR analysis, CASC9 was existed in both nucleus and cytoplasm of SNU-449 and HuH-6 cells (Figure 5(a)). In fluorescence in situ hybridization, we could observe that CASC9 was distributed in the nucleus and cytoplasm of SNU-449 cells (Figure 5(b)). Indeed, previous studies demonstrate that CASC9 is distributed in both nucleus and cytoplasm.16,19 Thus, CASC9 may function in the nucleus or cytoplasm to regulate gene expression at transcriptional or epigenetic level. Virtually, CASC9 has been reported to interact with EZH2 and subsequently regulate gene expression at epigenetic levels.28–31 RIP assay was used to evaluate the interaction between CASC9 and EZH2. We found that CASC9 was successfully pulled down by EZH2 compared with IgG control, indicating that CASC9 could bind to EZH2 directly (Figure 5(c)). Furthermore, CASC9 reduced the expression of PTEN in SNU-449 and HuH-6 cells (Figure 5(d)). In ChIP assay, knockdown of CASC9 by two short hairpin RNAs (sh-CASC9-1 and sh-CASC9-2) apparently reduced the occupancy of EZH2 to the promoter region of PTEN as well as H3K27me3 binding levels (Figure 5(e)). Besides, EZH2 was knocked out by two short guide RNAs (sg-EZH2-1 and sg-EZH2-2) and increased the expression of PTEN (Figure 5(f)). Collectively, we found that CASC9 epigenetically mediated PTEN expression by interacting with EZH2 in liver cancer cells.
Forced CASC9 expression partially abrogates propofol-induced AKT/mTOR inhibition in liver cancer cells
Phosphatase and tensin homolog is a well-known negative regulator of Akt/mTOR pathway. A number of studies report that propofol can suppress Akt/mTOR pathway in various cancers.32–34 Besides, CASC9 has been shown to activate Akt/mTOR signaling;16,35,36 thus, we speculated that propofol might suppress Akt/mTOR pathway partially by downregulating CASC9, and overexpression of CASC9 could counteract with propofol by activating Akt/mTOR pathway. In western blot analysis, propofol treatment reduced the expression levels of phosphorylated Akt and mTOR and increased the expression level of PTEN in SNU-449 and HuH-6 cells (Figure 6(a) and (b)). In contrary, CASC9 overexpression increased the expression levels of phosphorylated Akt and mTOR and reduced the expression level of PTEN in SNU-449 and HuH-6 cells (Figure 6(a) and (b)). Our results indicated that forced CASC9 expression partially abrogated propofol-induced AKT/mTOR inhibition in liver cancer cells.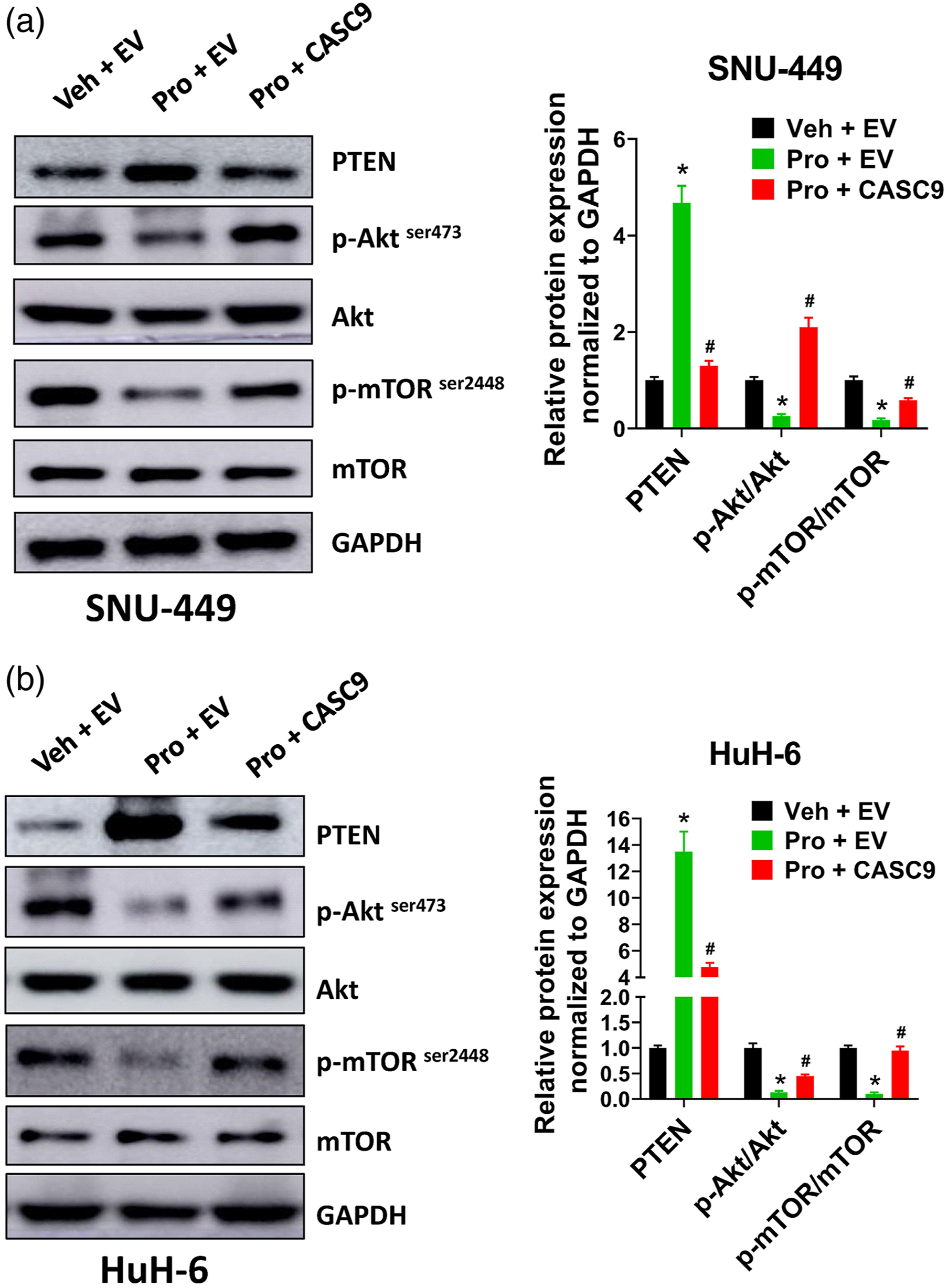
Discussion
Propofol is one of the most widely used anesthetic drugs in cancer surgery. In the present study, we found that propofol suppressed proliferation, migration, invasion, and tumor xenograft growth of liver cancer cells in vitro and in vivo. Our results provided preclinical evidence for using propofol-based anesthesia in liver cancer surgery and gave a possible explanation for the association of propofol and better survival of liver cancer patients. Indeed, there is increasing evidence suggest that propofol shows anti-cancer effects via multiple mechanisms. Propofol is reported to disrupt the cytoskeleton of cervical cancer cells, which may explain the influence of propofol on cell migration and invasion. 37 Propofol also shows anti-tumor effects in liver cancer. For example, a retrospective study of HCC patients compares the effects of propofol and inhalation anesthetics on postoperative recurrence and finds that propofol obviously reduces recurrence of HCC patients. 38 In HCC patients who undergoing hepatectomy, propofol anesthesia is associated with better survival compared with desflurane anesthesia. 7 Moreover, propofol suppresses growth and tumor xenograft formation of HepG2 cells by activating AMPK pathway and inducing autophagy. 39 Moreover, previous studies suggest that propofol exerts anti-tumor effects in various cancers by regulating the activation of signaling pathways and expression of protein-coding genes, microRNAs, and lncRNAs. 6 In our study, transcriptome RNA sequencing analysis was performed to identify dysregulated lncRNAs in propofol-treated liver cancer cells. We found that lncRNA CASC9 was significantly decreased by propofol, and forced expression of CASC9 partially abrogated the effects of propofol in liver cancer cells. Thus, our study provided a feasible strategy for searching lncRNAs that involved in propofol-mediated effects. Potential limitations for our study include that we did not know how propofol affected the expression of CASC9 in liver cancer cells, and the relation of structure of propofol and its anti-cancer effects and potential mechanisms.
CASC9 was upregulated in liver cancer tissues and associated with poor overall survival, indicating that CASC9 might exert oncogenic functions. In esophageal squamous cell carcinoma, CASC9 interacts with CBP in the nucleus and promotes metastasis by upregulating LAMC2. 16 CASC9 is overexpressed in oral squamous cell carcinoma and correlated with larger tumor size, lymph node metastasis, and overall survival. 36 Knockdown of CASC9 in oral squamous cell carcinoma cells promotes autophagy and apoptosis by suppressing Akt/mTOR pathway. In liver cancer, CASC9 is associated with cell viability and affects Akt signaling and DNA damage sensing of HCC cells. 35 CASC9 is overexpressed and acts as prognostic marker for HCC patients.40,41 In our study, propofol treatment reduced the expression of CASC9, and forced CASC9 expression partially abrogated the effects of propofol on cell growth, colony formation, invasion, and tumor growth of liver cancer cells. This might explain the inhibitory effect of propofol in liver cancer cells.
Our results indicated that propofol dose-dependently reduced the expression of exosomal CASC9 in liver cancer cells. In addition, exosomes from propofol-treated SNU-449 cells suppressed proliferation, migration, and invasion. Exosome is vital for establishing the microenvironment of liver cancer, and it also plays a role in the initiation, progression and metastasis. 42 Accumulated studies suggest that exosomal lncRNAs may act as diagnostic markers and promote progression and chemoresistance of cancer cells. For example, circulating lncRNA ATB from exosomes of HCC patients is upregulated and correlated with advanced TNM stage, larger tumor size, and poor overall survival. 43 Exosomal SENP3-EIF4A1 is dramatically decreased in HCC patients, and transfer of exosomal SENP3-EIF4A1 to HCC cells inhibits invasion, migration, and tumor growth. 44 In the present study, we proved that exosomes from propofol-treated SNU-449 cells exhibited anti-cancer effects, and CASC9 was downregulated by propofol in exosomes. Although connection between CASC9 and the inhibitory effects of exosomes from propofol-treated cells was ambiguous, our results provided a possible explanation for propofol-induced inhibitory effects.
In the present study, propofol treatment suppressed the activation of Akt/mTOR pathway. Indeed, previous studies demonstrate that multiple signaling pathways are influenced by propofol treatment, including Akt/mTOR pathway. 6 For instance, propofol suppresses migration, invasion, and PI3K/Akt pathway by inducing miR-206 expression and subsequently downregulating ROCK1. 32 In acute myeloid leukemia (AML), propofol induces apoptosis in AML differentiated cells and suppresses self-renewal of AML stem cells by inhibition Akt/mTOR and Wnt/β-catenin pathway. 45 Our results proved that propofol reduced the phosphorylation of Akt and mTOR and increased the expression level of PTEN, indicating that propofol suppressed Akt/mTOR pathway. As we discussed above, there is evidence that CASC9 exerts oncogenic functions by regulating Akt/mTOR signaling in a variety of cancers, including liver cancer.16,35,36 Our findings proved that CASC9 reduced PTEN expression by interacting with EZH2 and subsequently reduced H3K27me3 level at the promoter region of PTEN. Thus, Akt/mTOR signaling was activated by CASC9. We reasoned that the inhibitory effects of propofol on Akt/mTOR signaling were partially due to downregulating CASC9.
Conclusion
In summary, we found that propofol dose-dependently inhibited proliferation, migration, invasion, and tumor growth of liver cancer cells. Exosomes from propofol-treated SNU-449 cells also inhibited proliferation, migration, and invasion and promoted apoptosis. Transcriptional profiling of propofol-treated liver cancer cells identified lncRNA CASC9 as significantly downregulated lncRNA. Overexpression of CASC9 rescued the inhibitory effects of propofol on liver cancer cells. Furthermore, CASC9 was found to interact with EZH2 and reduced H3K27me3 binding at the promoter region of PTEN. Forced expression of CASC9 abrogated propofol-induced Akt/mTOR inhibition. Our results provided a novel insight into propofol-induced anti-tumor effects of liver cancer.
Supplemental Material
sj-pdf-1-het-10.1177_09603271211065972 – Supplemental Material for Propofol suppresses proliferation, migration, invasion, and tumor growth of liver cancer cells via suppressing cancer susceptibility candidate 9/phosphatase and tensin homolog/AKT serine/threonine kinase/mechanistic target of rapamycin kinase axis
Supplemental Material, sj-pdf-1-het-10.1177_09603271211065972 for Propofol suppresses proliferation, migration, invasion, and tumor growth of liver cancer cells via suppressing cancer susceptibility candidate 9/phosphatase and tensin homolog/AKT serine/threonine kinase/mechanistic target of rapamycin kinase axis by Qing Chang, Jun Wu, Yang An, Haiyan Liu and Yang Sun in Human & Experimental Toxicology
Supplemental Material
sj-xlsx-1-het-10.1177_09603271211065972 – Supplemental Material for Propofol suppresses proliferation, migration, invasion, and tumor growth of liver cancer cells via suppressing cancer susceptibility candidate 9/phosphatase and tensin homolog/AKT serine/threonine kinase/mechanistic target of rapamycin kinase axis
Supplemental Material, sj-xlsx-1-het-10.1177_09603271211065972 for Propofol suppresses proliferation, migration, invasion, and tumor growth of liver cancer cells via suppressing cancer susceptibility candidate 9/phosphatase and tensin homolog/AKT serine/threonine kinase/mechanistic target of rapamycin kinase axis by Qing Chang, Jun Wu, Yang An, Haiyan Liu and Yang Sun in Human & Experimental Toxicology
Footnotes
Authors' contributions
Yang Sun guaranteed the integrity of the entire study. The experiments were conducted by Yang Sun, Qing Chang, Jun Wu, and Yang An. Clinical studies were conducted by Haiyan Liu. Data were analyzed by Qing Chang. Manuscript was prepared and reviewed by Yang Sun. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Natural Science Foundation of Heilongjiang Province Joint Guide Project (LH2019H034), and Program for Innovation Research of Heilongjiang Provincial Hospital.
Ethical approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Heilongjiang Provincial Hospital (SYXK (Hei) 2015-013) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study. All procedures performed in studies involving animals were in accordance with the ethical standards of the ethics committee of the Heilongjiang Provincial Hospital.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.