
Abstract
The balance of cisplatin uptake and efflux, mediated mainly by organic cation transporter 2 (OCT2) and multidrug and toxin extrusion 1 (MATE1), respectively, determines the renal accumulation and nephrotoxicity of cisplatin. Using transporter-mediated cellular uptake assay, we identified wedelolactone (WEL), a medicinal plant-derived natural compound, is a competitive inhibitor of OCT2 and a noncompetitive inhibitor of MATE1. Wedelolactone showed a selectivity to inhibit OCT2 rather than MATE1. Cytotoxicity studies revealed that wedelolactone alleviated cisplatin-induced cytotoxicity in OCT2-overexpressing HEK293 cells, whereas it did not alter the cytotoxicity of cisplatin in various cancer cell lines. Additionally, wedelolactone altered cisplatin pharmacokinetics, reduced kidney accumulation of cisplatin, and ameliorated cisplatin-induced acute kidney injury in the Institute of Cancer Research mice. In conclusion, these findings suggest a translational potential of WEL as a natural therapy for preventing cisplatin-induced nephrotoxicity and highlight the need for drug–drug interaction investigations of WEL with other treatments which are substrates of OCT2 and/or MATE1.
Keywords
Introduction
Cisplatin (cis-diamminedichloroplatinum II) is a widely used chemotherapeutic agent for the treatment of various cancers, such as lung, ovarian, and breast cancers. 1 However, the clinical use of cisplatin is limited by its severe dose-dependent nephrotoxicity. 2 The kidney damage caused by the accumulation of cisplatin in renal cells mainly causes DNA damage, oxidative stress, inflammation, and apoptosis. 3 Currently, clinical treatments for alleviating or preventing cisplatin-induced nephrotoxicity frequently involve hydration, diuretics and magnesium supplementation; however, they may increase the risk of excessive diuresis and dehydration.4,5 Cytoprotective agents, such as amifostine and glutamine, are also used to reduce cisplatin toxicity, but they are usually accompanied by serious adverse reactions or unexpected tumor protection.6,7 Nonetheless, there is an urgent medical need for novel therapeutic options to prevent and/or treat cisplatin nephrotoxicity.
Organic cation transporter 2 (OCT2 or SLC22A2) is highly expressed in the basolateral membrane of the renal proximal tubule cells and mediates the transport of cationic compounds from the blood to the kidney. 8 Oct1/Oct2-deficient mice are resistant to cisplatin-induced renal tubular injury. 9 Furthermore, a non-synonymous single nucleotide polymorphism (SNP) in the OCT2 gene (rs316019) can also ameliorate the nephrotoxicity of cisplatin in humans. 10 Cimetidine (CIM) is an OCT2 inhibitor holding promise as a potential therapy for cisplatin-induced nephrotoxicity.11,12 Multidrug and toxin extrusion 1 (MATE1 or SLC47A1) is considered as the major transporter mediating the apical secretion of cisplatin. 13 Mate1 deficiency or selective MATE1 inhibitor potentiates nephrotoxicity of cisplatin in mice.14,15 Multidrug resistance-related protein 2 and 4 (MRP2/4) are membrane proteins in the MRP subfamily of ABC transporters, which are implicated in the removal of cisplatin conjugates from cells.16,17 Mrp2 deficiency reduces platinum excretion and aggravate kidney damage. 18 Therefore, the balance of uptake and efflux largely determines the renal accumulation and nephrotoxicity of cisplatin.
Wedelolactone (WEL) is a phenylpropanoid natural compound that has been used in clinical practice for the treatment of various disease, including infective hepatitis and snake venom poisoning.19,20 Additionally, WEL has been reported to demonstrate anti-tumor, anti-inflammatory anti-oxidant, anti-fibrosis, and anti-fatigue activities.21,22 However, it remains unknown about the effect of WEL on organic cation transport system in the kidney. The present study aimed to determine whether WEL can interact with the renal organic transport system and protect against cisplatin-induced acute kidney injury (AKI).
Materials and methods
Chemicals and reagents
Cisplatin was obtained from TCI Development Co. (Shanghai, China). Wedelolactone (98%) was bought from Chengdu Herb Purify (Chengdu, China). Sodium-diethyldithio-carbamate (DDTC) was bought from Tianjin Chemical Reagent Co. (Tianjin, China), and Nickel Chloride was purchased from Xilong Scientific Co. (Guangdong, China). 4-(4-(Dimethylamino) styryl)-N-methylpyridinium iodide (ASP+) was bought from Molecular Probes (Oregon, USA). Dulbecco’s modified Eagle’s medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640, and fetal bovine serum (FBS) were obtained from Biological Industries (BI, Galilee, Israel). Trypsin was obtained from Gibco (Maryland, USA). Methanol (HPLC Grade) was bought from Concord Technology Co. (Tianjin, China).
Cell culture
Human embryonic kidney 293 cells (HEK293) stably overexpressing empty vector (HEK-EV), human and mouse OCT2 (HEK-hOCT2, HEK-mOCT2) or MATE1 (HEK-hMATE1, HEK-mMATE1) were constructed according to our previous published method. 23 Briefly, the cDNAs of hOCT2, mOCT2, hMATE1, and mMATE1 were obtained from total RNA reverse transcription and sub-cloned into pcDNA3.1 plasmids (Invitrogen, Carlsbad, CA, USA). The recombinant plasmids were transfected into HEK293 cells using poly-ethylenimine (PEI) (Sigma-Aldrich, St. Louis, Missouri, USA). The empty pcDNA3.1 plasmid was transfected into HEK293 cells to establish control cells (HEK-EV cells). The cells were selected with hygromycin B (75 μg/mL) for 14 days. These cell lines were validated for their uptake function (Supplemental Figure S1). Human colon cancer cell line HCT116, breast cancer cell line MCF-7, and non-small cell lung cancer cell line A549 were frozen in our laboratory; human prostate carcinoma cell line DU145 was a gift from Professor Dexin Kong (Tianjin Medical University, Tianjin, China). The cells were cultured in a complete DMEM medium (DU145 in RPMI 1640 complete medium) containing 10% FBS, 1% penicillin/streptomycin at 37°C, and 5% CO2. For HEK293 overexpressing cells, 75 μg/mL hygromycin B was added.
Uptake experiments in HEK293 cells
HEK293 cells overexpressing the indicated transporter (4 × 104/well) were seeded on the 96-well cell culture plates coated with poly-D-lysine (Sigma-Aldrich, St. Louis, Missouri, USA) for 24 h before performing the uptake experiments. For the OCT2-mediated uptake experiment, cimetidine was used as a prototypical inhibitor of OCT2, and ASP+ was used as a fluorescent substrate of OCT2. Cells were incubated with pre-warmed OCT2 uptake buffer (137.9 m
Cell viability assay
To determine the non-toxic concentrations of WEL in kidney cells, three cell lines (HEK-EV, HEK-hOCT2, and HEK-mOCT2) were incubated with WEL (0.5, 1, 2 or 5 μM) for 24 h. To determine whether WEL-altered cisplatin toxicity in kidney cells via interaction with OCT2, HEK-hOCT2, and HEK-mOCT2 cells (1.0 × 104 cells/well) were treated with cisplatin (2 μM), WEL (1 μM) or cisplatin (2 μM) +WEL (1 μM) for 24 h. To determine whether WEL-altered cisplatin anti-tumor activity in cancer cells, various cancer cells lines (8 × 103 cells/well) were treated with serially diluted cisplatin (0, 1, 5, 10, 15, 20, 50, and 100 μM) in the presence or absence of WEL (0, 1 and 5 μM) for 48 h. The cell viability was determined by using the MTT assay. After treatment, MTT reagent was added to the cells and incubated at 37°C for 4 h. The absorbance was measured at 490 nm wavelength on the microplate reader. 24
Animal study
Male Institute of Cancer Research (ICR) mice (6–7 weeks old, 32–34 g) were purchased from Vital River Laboratory Animal Technology (Beijing, China), and housed in a specific pathogen-free environment (temperature: 23 ± 2°C, humidity: 60 ± 5%, 12 h dark/light cycles) in the laboratory animal center of the Institute of the Radiation Medicine Chinese Academy of Medical Sciences (CAMS, Tianjin, China) with water and food ad libitum. The number of the animal use permit is SYXK2019-0002. Before starting the study, the animal research protocol was reviewed and approved by the Animal Ethical and Welfare Committee (AEWC) at the CAMS (approval number: IRM-DWLL2020127). In addition, all experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals.
To determine the pharmacokinetics (PK) of cisplatin, ICR mice (n = 4/group) were orally treated with vehicle solution (20% PEG400 in 0.5% sodium carboxymethyl cellulose, p.o.) or WEL (20 mg/kg) 30 min before an intraperitoneal injection (i.p.) of cisplatin (20 mg/kg). The doses of cisplatin and WEL were selected according to both our preliminary study and literature. In our preliminary study, we tried several doses of cisplatin and WEL for toxicity and PK. We found that a single dose of 20 mg/kg cisplatin was able to induce AKI in mice after 72 h, which is consistent with previous literature.12,25,26 Additionally, we also found that 20 mg/kg WEL was able to significantly increase plasma levels of cisplatin in our preliminary study. The blood was obtained by orbital bleeding at 5, 15, 30, 45, 60, and 90 min after cisplatin injection. The blood was collected into tubes moistened with heparin and centrifuged at 3000 r/min for 15 min to obtain plasma samples.
To determine the nephrotoxicity of cisplatin in the presence of WEL, mice were divided into three groups (n = 5–6/group): control group (Cont), cisplatin group (CIS), and cisplatin + WEL group (CIS + WEL). Mice were orally treated with vehicle solution (20% PEG400 in 0.5% sodium carboxymethyl cellulose) or WEL (20 mg/kg) 30 min before a single dose of saline or cisplatin (20 mg/kg). The dose was chosen according to previous studies.27–29 After 72 h administration of cisplatin, mice were anesthetized to collect blood and kidneys. Blood urea nitrogen (BUN) and creatinine (CRE) were measured using commercial kits (Nanjing Jiancheng Biological Technology, Nanjing, China).
Histopathology
The right kidneys were immediately cut longitudinally and fixed in 4% neutral-buffered paraformaldehyde for 48 h. Kidney tissues were then dehydrated by passing through graded concentrations of ethanol and cleared by passing through xylene. After embedding in paraffin, kidney tissues were cut into sections with a thickness of 5 μm. The sections were then deparaffinized with xylene, rehydrated with graded concentrations of ethanol, and stained with hematoxylin and eosin (H&E). The kidney sections were examined under light microscope by Prof. Maojuan Guo, a pathologist at Tianjin University of Traditional Chinese Medicine (Tianjin, China; Email:
RNA extraction and RT-qPCR analysis
Total RNA of kidneys was extracted using Trizol reagent (Takara, Dalian, China) according to the manufacturer’s instructions. Total RNA (800 ng) was used to synthesize cDNA with HisScript QRT SuperMix (Vazyme Biotech, Nanjing, China), a high-efficiency cDNA first-strand synthesis kit. Ultra SYBR Mixture (Cwbio Bio Inc., Beijing, China) was used to quantify the mRNA expression of the target genes on an ABI QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems, Foster City, CA). The fold change in various mRNAs was calculated according to the comparative threshold cycle (Ct) method (2−ΔΔCt). Mouse Gapdh was used as the housekeeping gene for normalization. The primer sequences of the genes are listed in Supplemental Table S1.
Quantification of cisplatin in mouse plasma
HPLC-UV was used to quantitate the cisplatin in mouse plasma as described previously with minor modifications.33–35 Briefly, a C18 reversed phase column (150 mm × 4.6 mm, 5 μm) was used for the separation on an Agilent 1260 HPLC (Agilent Technologies, USA) at a column temperature of 35°C. The optimized mobile phase was water: methanol at 28:72 with a flow rate of 1.1 mL/min. The absorption wavelength was set at 254 nm. The total run time for the assay was 10 min.
The stock solution of cisplatin (1 mg/mL) was prepared in 0.9% NaCl and diluted into calibrators using blank plasma. Nickel chloride (1 mg/mL) was dissolved in 0.9% NaCl as the internal standard (IS). DDTC (10%) was dissolved in 0.1
Statistical analysis
The data were statistically analyzed using GraphPad Prism software, version 7.00 and expressed as mean ± SEM. Two-tailed unpaired Student’s t-test was used to analyze the differences between two groups. The differences between more than two groups were analyzed using a one-way analysis of variance (ANOVA) followed by two-by-two comparisons using Tukey’s test. p < .05 is considered significant.
Results
Inhibitory effect of wedelolactone on OCT2
The inhibitory effect of WEL on both human and mouse OCT2 was evaluated by determining the effect of gradually diluted WEL on the accumulation of ASP+ in HEK-EV, HEK-hOCT2 and HEK-mOCT2 cells. Cimetidine (CIM) has been used as a prototypical OCT2 inhibitor to protect cisplatin-induced kidney injury.
11
Thus, we used CIM as the positive control. As shown in Figure 1(a), WEL inhibited both human and mouse OCT2-mediated ASP+ uptake in a concentration-dependent way. The calculated IC50 values of WEL and CIM were 19.14 μM and 57.22 μM for human OCT2, as well as 12.28 μM and 47.20 μM for mouse OCT2, respectively. In Lineweaver–Burk plots, WEL and CIM showed a competitive inhibition of both human OCT2 and mouse OCT2 (Figure 1(b)). These results suggest that WEL is a competitive inhibitor of both human and mouse OCT2. Inhibitory potency and mechanisms of wedelolactone for human and mouse OCT2. (a) The inhibitory effect of wedelolactone (WEL) and cimetidine (CIM) (0–1000 μM) on the accumulation of ASP+ (5 μM) in HEK-hOCT2 and HEK-mOCT2 cells. The X-axis is the logarithmic value of WEL or CIM concentration. (b) The inhibitory mechanism of different concentrations of WEL (orange) and CIM (blue) on OCT2-overexpressing cells was measured at the increasing ASP+ concentration. The X- and Y-axis represent the reciprocal of ASP+ concentration and fluorescence intensity, respectively. Data were expressed as mean ± SEM, n = 4.
Inhibitory effect of wedelolactone on MATE1
Studies have shown that OCT2 and MATE1 have substrate cross-specificity and share a broad range of substrates.36–38 We next explored whether WEL interacts with human and mouse MATE1. A concentration-dependent decrease of ASP+ specific uptake was observed in both HEK-hMATE1 and HEK-mMATE1 cells after treating with WEL or CIM at increasing concentrations (Figure 2(a)). The IC50 values of WEL and CIM were 23.51 μM and 19.77 μM for human MATE1, as well as 46.85 μM and 7.01 μM for mouse MATE1, respectively. Lineweaver–Burk plots showed that WEL and CIM noncompetitively inhibited both human and mouse MATE1 (Figure 2(b)). These results suggest that WEL is a noncompetitive inhibitor of both human and mouse MATE1. Inhibitory potency and mechanisms of wedelolactone for human and mouse MATE1. (a) The inhibitory effect of wedelolactone (WEL) and cimetidine (CIM) (0–1000 μM) on the accumulation of ASP+ (5 μM) in HEK-hMATE1 and HEK-mMATE1 cells. The X-axis is the logarithmic value of WEL or CIM concentration. (b) The inhibitory mechanisms of WEL (orange) or CIM (blue) on MATE1-overpressing cells. The X- and Y-axis represent the reciprocal of ASP+ concentration and fluorescence intensity, respectively. Data were expressed as mean ± SEM, n = 4.
Effect of wedelolactone on cell viability of cisplatin-treated cells
To determine the non-toxic concentrations of WEL in kidney cells, three cell lines (HEK-EV, HEK-hOCT2, and HEK-mOCT2) were incubated with WEL at the indicated concentrations for 24 h. As shown in Figure 3(a), WEL at >1 μM slightly inhibited the growth of both HEK-hOCT2 and HEK-mOCT2 cells. Due to low expression of OCT2, HEK-EV cells were more resistant to cisplatin-induced cytotoxicity compared with HEK-hOCT2 or HEK-mOCT2 cells (data not shown). Therefore, 1 μM WEL was chosen for the subsequent cellular experiments. To determine whether WEL-altered cisplatin toxicity in kidney cells via interaction with OCT2, HEK-hOCT2, and HEK-mOCT2 cells were treated with the indicated concentrations of cisplatin or WEL for 24 h. As shown in Figure 3(b), cisplatin significantly inhibited the cell growth of both HEK-hOCT2 and HEK-mOCT2 cells. In contrast, co-treatment of WEL almost completely reversed cisplatin-induced cytotoxicity. The cell morphology confirmed that WEL had a protective effect on the cisplatin-induced cytotoxicity in HEK-hOCT2 and HEK-mOCT2 cells (Figure 3(c)). Taken together, WEL protects against cisplatin-induced cytotoxicity in OCT2-overexpressing HEK293 cells. Effect of wedelolactone on cell viability of cisplatin-treated cells. (a) HEK-hOCT2 and HEK-mOCT2 cells were incubated with WEL at the indicated concentrations for 24 h. Cell viability was determined by MTT assay. *p < .05, **p < .01 vs. control cells (0 μM WEL). Data were expressed as mean ± SEM, n = 4. (b and c) HEK-hOCT2 and HEK-mOCT2 cells were treated with 2 μM cisplatin (CIS), 2 μM cisplatin+ 1 μM wedelolactone (CIS + WEL), or 1 μM wedelolactone (WEL) for 24 h. Cell viability was determined by MTT assay (b). *p < .05, **p < .01 vs. Cont group, #p < .05 vs. CIS group. Data were expressed as mean ± SEM, n = 4. Representative images of cell morphology (magnification ×200, scale bar: 100 μm) (c).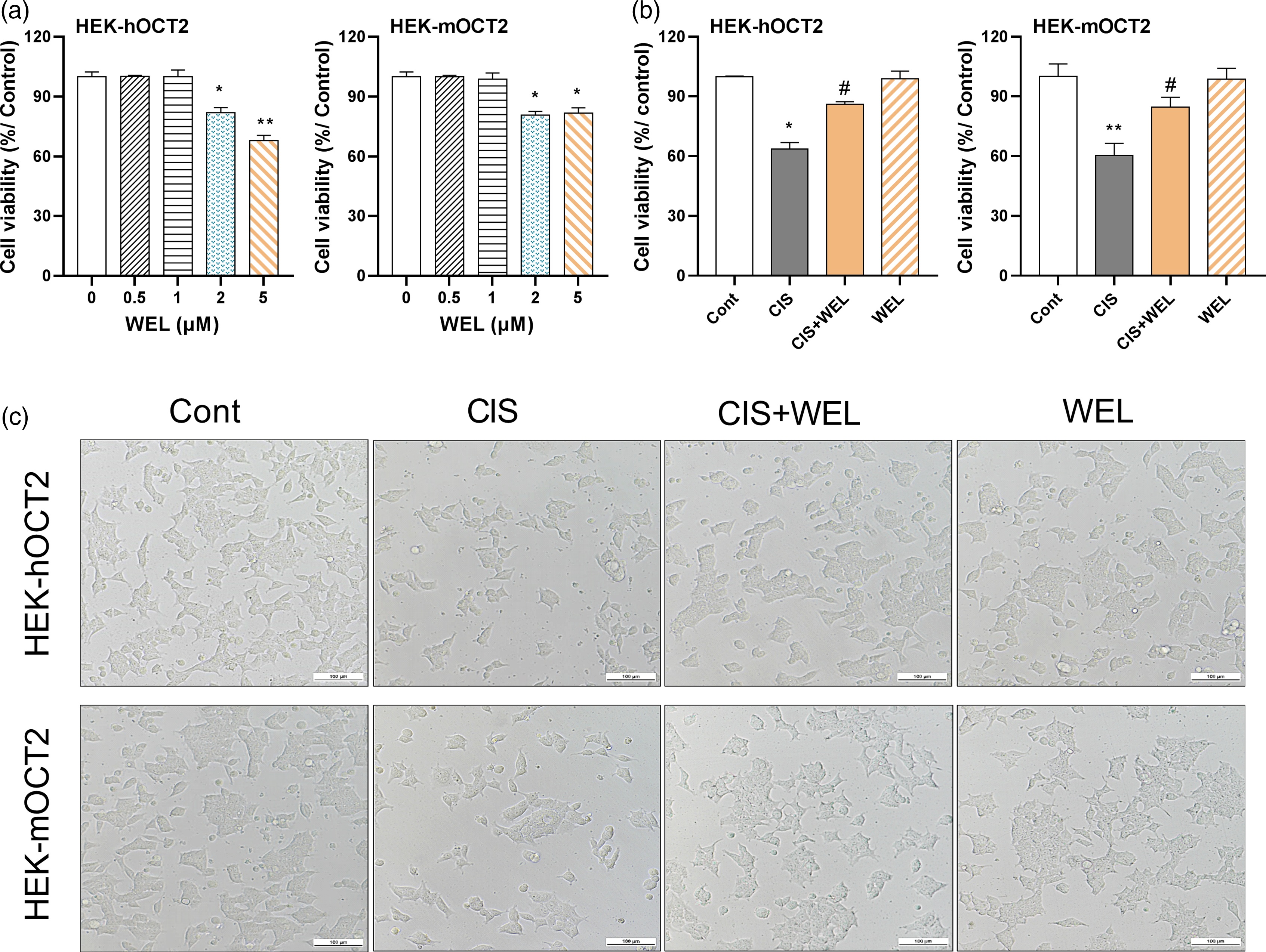
Wedelolactone altered the pharmacokinetics of cisplatin in mice
In our preliminary study, mice were administered two doses (20 and 100 mg/kg) of WEL. We observed that WEL at 20 mg/kg significantly increased the plasma concentration of cisplatin (data not shown). Therefore, 20 mg/kg WEL was chosen to investigate its effect on the PK of cisplatin. As shown in Figure 4, WEL significantly increased plasma concentrations of cisplatin compared to CIS group. The area under the curve (AUC0–90 min) of cisplatin was also significantly higher in WEL co-treatment group than CIS group (826.2 ± 16.8 μg·min/mL vs. 656.0 ± 30.2 μg·min/mL, p = .043). These results suggested that WEL increases the systemic exposure of cisplatin in mice. Effect of wedelolactone on the plasma cisplatin levels in mice. ICR mice were administrated with vehicle solution (20% PEG400 in 0.5% sodium carboxymethyl cellulose) or WEL (20 mg/kg) orally 30 min before intraperitoneal injection (i.p.) of cisplatin (20 mg/kg). The plasma samples were obtained at 5, 15, 30, 45, 60, and 90 min after cisplatin injection for HPLC analysis. Mean ± SEM from four mice/group was shown. *p < .05, **p < .01 vs. CIS group.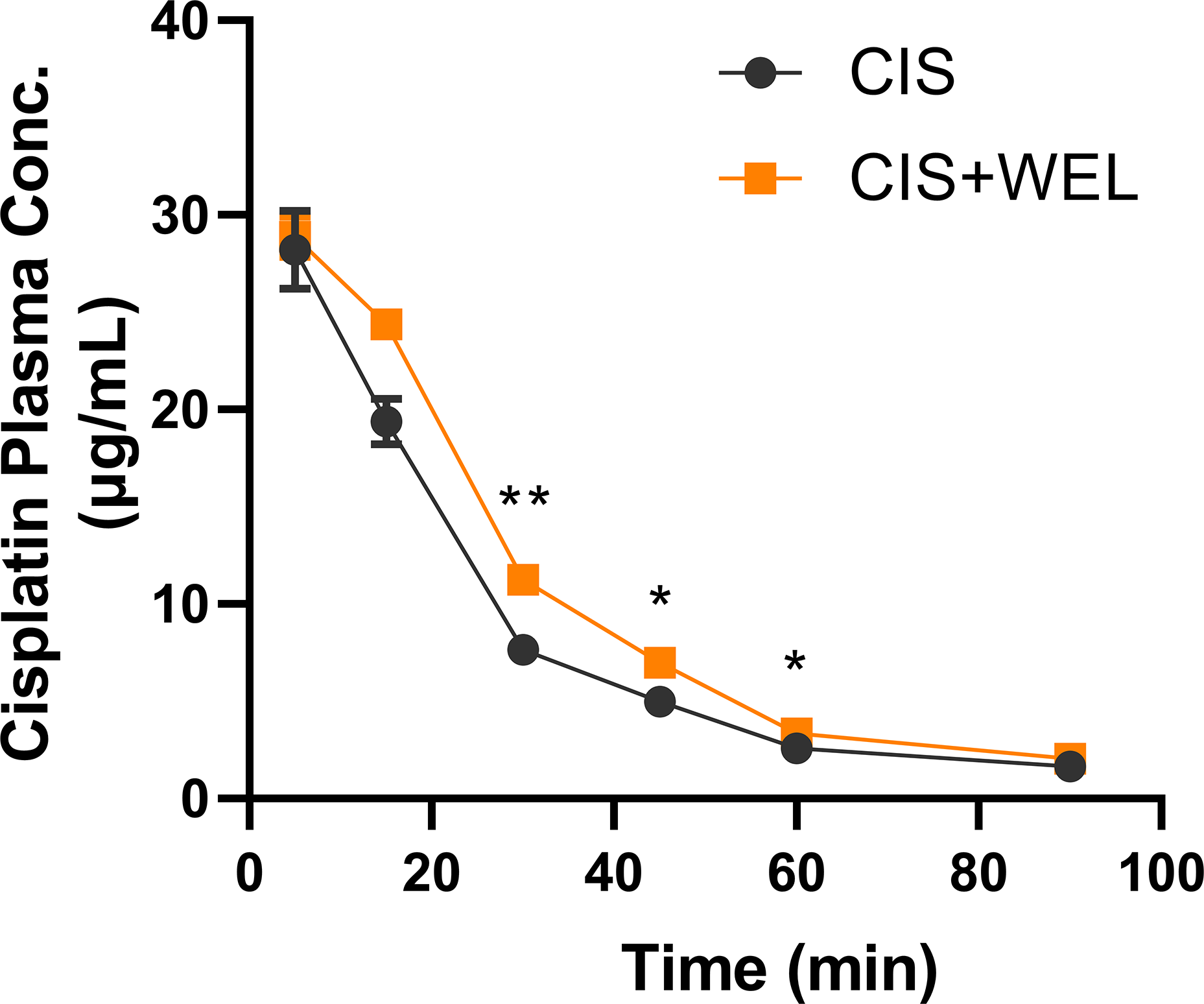
Wedelolactone alleviated cisplatin-induced acute kidney injury in mice
Mice were treated with WEL or vehicle by gavage 30 min before cisplatin administration to study the effect of WEL on AKI caused by cisplatin (Figure 5(a)). The significant increase in plasma BUN and CRE proved that 20 mg/kg cisplatin for 72 h could obviously cause kidney damage in mice (Figures 5(b) and (c)). In contrast, co-treatment of WEL significantly decreased plasma BUN and CRE in cisplatin-treated mice. Protective effect of wedelolactone on cisplatin-induced nephrotoxicity in mice. (a) Male ICR mice were divided into 3 groups (n = 5–6/group): control group (Cont), cisplatin group (CIS), and cisplatin + WEL group (CIS + WEL). Mice were orally treated with vehicle solution (20% PEG400 in 0.5% sodium carboxymethyl cellulose) or WEL (20 mg/kg) 30 min before a single dose of saline or cisplatin (20 mg/kg). Blood and kidney tissue were obtained 72 h after cisplatin injection. (b) Blood urea nitrogen (BUN). (c) Plasma creatinine (CRE). (d) Representative H&E staining of mouse kidneys (magnifications ×200 and ×400, scale bar: 100 μm). Black solid arrowheads represent tubular cells necrosis, black hollow arrowheads indicate tubule dilatation and protein cast formation, and red boxes represent inflammatory cell infiltration. (e) Analysis of tubular injury score. Data were shown as means ± SEM of 10 random fields from each mouse. (f) Kidney/body weight ratios. (g) Relative Kim-1 mRNA expression in mouse kidneys. (h) Kidney cisplatin accumulation after 72 h treatment measured by HPLC. Data were expressed as mean ± SEM, n = 5–6. **p < .01, ***p < .001 vs. Cont group; #p < .05, ##p < .01 vs. CIS group.
In the macroscopic examination (please see inset figures of Figure 5(d) at bottom left corners), it was noted that the kidneys of the mice treated with cisplatin were paler compared to the control group. H&E staining showed that cisplatin induced significant morphological alterations in mouse kidneys, including tubular cell necrosis, tubular dilatation, protein cast formation, and inflammatory cell infiltration in the interstitial area (Figure 5(d)). Such abnormalities, especially cell necrosis and tubular dilatation in kidneys of cisplatin-treated mice, were markedly alleviated by co-treatment of WEL (Figures 5(d) and (e)). There was still inflammatory cell infiltration in kidneys of CIS+WEL group, but it was greatly reduced compared with kidneys of CIS group where massive inflammatory cell infiltration was observed. Additionally, co-treatment of WEL also significantly decreased kidney/body weight ratios and Kim-1 mRNA levels in kidneys of cisplatin-treated mice (Figures 5(f) and (g)). Notably, a lower renal accumulation of cisplatin was found in WEL co-treatment group compared with cisplatin group (Figure 5(h)). These results indicate that WEL prevents cisplatin-induced renal injury and reduces renal accumulation of cisplatin in mice.
Effect of wedelolactone on mRNA expression of genes involved in cisplatin transport and cisplatin-induced inflammation and oxidative stress
The kidneys of cisplatin-treated mice underwent adaptive responses to kidney injury by altering the expression of transporters involved in cisplatin accumulation. As shown in Figure 6(a), cisplatin markedly decreased the mRNA expression of Oct2 and Mate1, whereas it significantly increased the mRNA expression of Mrp2 and Mrp4 in mouse kidneys. Such alterations were significantly reversed by co-treatment of WEL, except for Mrp2. As shown in Figure 6(b), cisplatin markedly up-regulated the mRNA expression of two inflammatory biomarkers, namely, tumor necrosis factor alpha (Tnfα) and inducible nitric oxide synthase (iNos), as well as two oxidative stress biomarkers, namely, quinone dehydrogenase 1 (Nqo1) and heme oxygenase-1 (Ho-1). Notably, WEL markedly decreased the mRNA expression of iNos, Nqo1, and Ho-1 in kidneys of cisplatin-treated mice. Effect of wedelolactone on mRNA expression of genes involved in cisplatin transport and cisplatin-induced inflammation and oxidative stress. Male ICR mice were divided into 3 groups (n = 5–6/group): control group (Cont), cisplatin group (CIS), and cisplatin + WEL group (CIS + WEL). Mice were orally treated with vehicle solution (20% PEG400 in 0.5% sodium carboxymethyl cellulose) or WEL (20 mg/kg) 30 min before a single dose of saline or cisplatin (20 mg/kg). (a) The mRNA expression of transporters in kidneys. (b) The mRNA expression of inflammatory and oxidant genes. Gapdh was the housekeeping gene. Relative mRNA levels were calculated as fold change normalized to controls. Data were expressed as mean ± SEM, n = 5–6. *p < .05, **p < .01, ***p < .001 vs. Cont group; #p < .05, ##p < .01 vs. CIS group.
Wedelolactone had no effect on cytotoxicity of cisplatin in cancer cell lines
To determine whether the WEL alters the anti-tumor activity of cisplatin, we determined the effect of WEL on cisplatin-induced cytotoxicity in HCT116, A549, MCF-7, and DU145, respectively. As shown in Figure 7 and Supplemental Table S2, WEL (1 and 5 μM) had no significant effect on the 50% growth inhibition concentration (GI50) of cisplatin in these cancer lines. Effect of wedelolactone on anti-tumor activity of cisplatin in cancer cells. HCT116, A549, MCF-7 and DU145 cells were treated with the indicated concentrations of cisplatin combined with 0 μM, 1 μM, and 5 μM WEL for 48 h. Cell viability and GI50 values were calculated, and the curves were obtained by nonlinear regression fitting by GraphPad 7.00. Data were expressed as mean ± SEM, n = 4.
Discussion
The renal accumulation and nephrotoxicity of cisplatin are determined by the balance of its uptake and efflux in the kidney. Cisplatin uptake in the kidney is mainly mediated by OCT2, which is highly expressed in the basolateral membrane of the kidney. 39 Cimetidine has been used as the prototypical OCT2 inhibitor to investigate the role of OCT2 in cisplatin-induced nephrotoxicity. 40 Since then, numerous studies have shown the protective effect of OCT2 inhibitors on cisplatin nephrotoxicity. In contrast to the basal uptake transporter OCT2, the apical efflux transporter MATE1 is mainly responsible for cisplatin excretion into urine. 13 Pyrimethamine as a MATE1 inhibitor significantly enhance cisplatin-induced nephrotoxicity in mice. 14 Notably, OCT2 and MATE1 are both multi-specific organ cation transporters that share an overlapping substrate spectrum. It is not surprising that cisplatin-induced nephrotoxicity could be alleviated by low doses of OCT2 inhibitor, but significantly aggravated by high doses of OCT2 inhibitor due to non-selective inhibition of MATE1. 41 Although cimetidine showed protective effect on cisplatin-induced nephrotoxicity, our data showed that it is a more potent inhibitor of MATE1 than OCT2, which is consistent with previous literature. 42 This may also suggest that OCT2 plays a more important role in regulating cisplatin accumulation and nephrotoxicity than MATE1. Ideally, a selective OCT2 inhibitor with no inhibitory effect on MATE1 is highly desired to protect against cisplatin-induced nephrotoxicity.
Wedelolactone is a phenylpropanoid natural compound with diverse biological activities. For instance, WEL alleviates doxorubicin-induced renal damage by reducing inflammation and oxidative stress via IκK/IκB/NF-κB pathway. 43 Miao et al. reported that WEL protects against cisplatin-induced renal injury via inhibition of caspase-11. 29 Although WEL has been used in clinical practice for a long history, it remains largely unknown whether WEL alters the function of renal drug transporters. Considering the importance of OCTs and MATEs in transporting organic cation drugs, it is of clinical significance to investigate the interaction of WEL with these transporters and to predict the potential drug–drug interactions. To the best of our knowledge, we identify for the first time that WEL is an inhibitor of both OCT2 and MATE1, showing higher selectivity toward OCT2 than cimetidine and showing a specificity toward OCT2 versus MATE1. The IC50 ratio (OCT2/MATE1) of WEL was 0.8 for humans and 0.3 for mouse, respectively. Therefore, the effect of WEL on the reduction of cisplatin accumulation is mainly achieved by inhibiting OCT2.
In the present study, a HPLC-UV method was established to determine the concentration of free cisplatin in mouse plasma and tissues. It is reported that about 90% of cisplatin is bound to plasma proteins or other macromolecules, while only 10% is free. 44 Additionally, plasma cisplatin levels decline significantly within 1 h of administration with a plasma half-life of 25–49 min28,45 Our preliminary study showed that it is difficult to detect plasma-free cisplatin using HPLC-UV after 2 h of cisplatin injection. Thus, we performed a 90-min PK study of cisplatin in the present study. WEL could significantly increase plasma cisplatin levels at 30, 45, and 60 min. It should be noted that the present PK study is limited by only one dose level of WEL and the HPLC-UV method. Future studies are needed to include multiple doses of WEL and use ICP-MS for the cisplatin quantification in the PK study of cisplatin.
In this study, a single intraperitoneal injection of cisplatin (20 mg/kg) is able to induce AKI, as evidenced by elevated plasma levels of BUN and CRE, increased kidney/body weight ratios, as well as whitening kidneys. Particularly, H&E analysis revealed prominent tubular cell necrosis, tubular dilatation, cast formation, and inflammatory cell infiltration in kidneys of cisplatin-treated mice, which is consistent with the observations in previous studies.3,29,46 Such alterations were significantly reversed by WEL co-treatment. Additionally, WEL shows anti-inflammatory and anti-oxidant activities in cisplatin-treated mice, as evidenced by decreased expression of genes involved in inflammation and oxidative stress. However, it remains unclear whether anti-inflammation and anti-oxidant effects of WEL are a result or a cause of its protection. Nonetheless, we have enough evidence that WEL is an OCT2 inhibitor and cisplatin is a substrate of OCT2. Notably, a recent study showed that Oct2 deficiency protects against cisplatin nephrotoxicity in mice partly because OCT2 is an active regulator of the renoprotection protein network, including proinflammatory and profibrotic proteins. 47 Future studies using Oct2−/− mice and omics technologies are required to decipher the exact underlying mechanisms for the protective effect of WEL on cisplatin-induced nephrotoxicity.
Although inhibitors of OCT2 and MATE1 have been shown to modulate cisplatin accumulation and nephrotoxicity, it should be noted that the expression of transporters is significantly altered by cisplatin-induced AKI. Previous studies reported that renal levels of OCT2 were markedly down-regulated (>70%↓) in both rats and mice after cisplatin treatment. 48 Despite unaltered expression of MATE1, the expression of other efflux transporters, such as Mrp2 and Mrp4, was significantly increased in kidneys of cisplatin-treated mice. 17 Notably, both in vitro and in vivo studies suggest that MRP2 mediates the efflux of cisplatin from kidney and regulates the renal accumulation of cisplatin. 18 Therefore, enhanced expression of efflux transporters (MRPs) along with concomitant decreases in uptake transporters (OCT2) may serve a defense mechanism to promote tubule cell survival. In the present study, we showed that cisplatin markedly decreased the mRNA expression of Oct2 and Mate1, but significantly increased the mRNA expression of Mrp2 and Mrp4 in the kidney. These alterations could be alleviated by co-treatment of WEL. It may seem to be contradictory that WEL, an OCT2 inhibitor, protects against nephrotoxicity in cisplatin-treated mice with a prominent down-regulation of Oct2 mRNA expression. It should be noted that WEL could prevent renal accumulation of cisplatin via inhibition of OCT2 in mice at the early stage of cisplatin treatment when OCT2 protein remains at the normal level. The lower cellular accumulation of cisplatin leads to less down-regulated OCT2 in the kidney. It should be also noted that only male mice were used in the present study. Both human and murine kidneys show gender-specific differences in response to injury. 49 Additionally, in both mice and rats, Oct2 mRNA expression is higher in males than females.50,51 In the future, we will focus on the gender-different effect of OCT2 inhibitors on cisplatin-induced kidney injury.
In summary, we showed that WEL could ameliorate cisplatin-induced nephrotoxicity at least partially by reducing the accumulation of cisplatin in renal tubular cells through inhibition of OCT2 uptake activity. WEL also shows anti-inflammatory and anti-oxidant activities in cisplatin-treated mice. Additionally, the present study provides a first evaluation of the relevance to investigate a clinical drug–drug interaction between WEL and cisplatin via the inhibitory effect of WEL on OCT2 and MATE1. These findings highly suggest a translational potential of WEL as a natural therapy for preventing cisplatin-induced nephrotoxicity and highlight the need for drug–drug interaction investigations of WEL with other treatments which are substrates of OCT2 and/or MATE1.
Supplemental Material
sj-pdf-1-het-10.1177_09603271211047915 – Supplemental Material for Wedelolactone protects against cisplatin-induced nephrotoxicity in mice via inhibition of organic cation transporter 2
Supplemental Material, sj-pdf-1-het-10.1177_09603271211047915 for Wedelolactone protects against cisplatin-induced nephrotoxicity in mice via inhibition of organic cation transporter 2 by Guangju Wang, Yajuan Bi, Hui Xiong, Tongwei Bo, Lifeng Han, Lijun Zhou, Chunze Zhang and Youcai Zhang in Human & Experimental Toxicology
Footnotes
Author contributions
YZ and GW: conceptualization and research design; GW, YB, HX, and TB: perform the experiments; GW, YB, CZ, LZ, LH, and YZ: analysis and interpretation of data; YZ, CZ, and GW: lead the research, writing/editing the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Key R&D Projects in the Tianjin Science and Technology Pillar Program (No. 19YFZCSY00420), the Project of Innovation Foundation of Tianjin University-Qinghai Nationalities University (2020XGP-0074), and the National Natural Science Foundation of China (No. 81673523). This work was also supported by grants from the Natural Science Foundation of Tianjin City (No. 17JCZDJC33000), and the National Key R&D Program of China (No. 2017YFC1700606 and No. 2017YFC1700604), and the National Key R&D Program of China (No. 2017YFC1700606 and No. 2017YFC1700604).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.