
Abstract
Erythropoietin (EPO) has antiapoptotic, antioxidative, and anti-inflammatory effects on ischemia tissues and protects against acute lung injury (ALI) induced by ischemia-reperfusion (I/R). p38 mitogen-activated protein kinases (p38 MAPK) signaling is involved in the processes of I/R-induced ALI. However, the interaction of EPO with p38 MAPK signaling in I/R-induced ALI has not been reported. To explore this issue, we constructed an I/R-induced ALI model in vivo and in vitro using Sprague Dawley rats and BEAS-2B cells. Some I/R rats and hypoxia-reoxygenation (H/R)–induced cells were treated with EPO, and the others were used as control groups. The injuries of lung tissues and cells were respectively assessed by inflammatory cytokine, morphologic changes, cell viability, apoptosis, and oxidative damage–related factors. Western blot determined key proteins in the p38 MAPK signaling. Results indicated that I/R induced the increase of inflammatory factors, lung weight, filtration coefficient, bronchoalveolar lavage fluid protein content, apoptosis, neutrophil, and lung peroxidation, and H/R caused cell growth inhibition, apoptosis, and oxidative damage-related factors’ release. EPO attenuated I/R-induced injury in vivo and in vitro. Furthermore, the increase of p-p38, p-JNK, and p-ERK1/2 in lung tissues and cells induced by I/R was downregulated by EPO. Moreover, both EPO and an inhibitor of p38 MAPK (SB203580) alleviated H/R-induced cell injury. Erythropoietin along with SB203580 had more obvious protection effects than EPO alone. Collectively, EPO alleviated I/R-induced ALI by blocking p38 MAPK signaling. The interaction mechanism of EPO with p38 MAPK signaling contributes to understanding the processes of I/R-induced ALI and provides new insights for the disease treatment.
Introduction
Acute lung injury (ALI) induced by ischemia-reperfusion (I/R) occurs when ventilation and/or blood supply is restored to ischemic lung tissue. 1 Although I/R can maintain cell viability and restore tissue function, I/R also causes organ damage and injury by triggering a cascade of biochemical changes. 2 The process of I/R-induced ALI involves the generation of reactive oxygen species (ROS) and the activation of the immune response and pulmonary alveolar macrophages. 3 As a result, I/R-induced ALI leads to lung edema, nonspecific alveolar damage, and vascular permeability, as well as neutrophil sequestration, 4 which frequently develops in many clinical conditions, mainly including lung transplantation, septic shock, pulmonary embolism, and cardiopulmonary bypass. 5 Nevertheless, the specific molecular mechanisms related to the process of I/R-induced ALI are still not fully studied.
Erythropoietin is a hematopoietic growth factor, mainly produced by the adult kidney and fetal liver in response to hypoxia, which is critical for regulating the production of red blood cells. 6 EPO has been applied for many years to treat anemia that results from different diseases such as chronic renal failure, prematurity, and cancer. 7 In addition, EPO has been reported to have antiapoptotic, antioxidative, and anti-inflammatory properties8–10 and consequently protects against I/R injury in various tissues, mainly including kidney, 11 heart, 12 and brain. 13 Moreover, the protective effect of EPO has also been found in different lung injuries. 14 In acute necrotizing pancreatitis-induced ALI, EPO can reduce oxidative stress, preserve microvascular endothelial cell integrity, and decrease proinflammatory cytokines’ levels. 15 EPO downregulates the proinflammatory levels and reduces apoptosis as well as oxidative stress to attenuate ALI induced by seawater aspiration. 16 EPO suppresses the expression of TNF-α and MMP-9 to protect against lung I/R injury. 17 However, the detailed cellular and molecular signaling pathways involved in the protective effect of EPO in I/R-induced ALI remain unclear.
p38 mitogen-activated protein kinases (p38 MAPK, p38) are a class of evolutionarily conserved serine/threonine MAPKs that regulate many cellular processes by linking extracellular signals to the intracellular machinery. 18 The p38 MAPK family comprises four kinases (p38α, β, γ, and δ). 19 The p38 MAPK signaling pathway is activated in response to different cellular and environmental stresses, inflammatory stimuli, and other signals, thereby regulating cell survival, differentiation, apoptosis, and autophagy.19,20 The p38 MAPK pathway has been considered as a potential therapeutic target for various diseases (e.g., cancer, diabetic cardiomyopathy, and chronic pain).21–23 Moreover, Chen et al. have shown that the activation of p38 MAPK pathway is related to TNF-α and IL-β and aggravates lung injury induced by burn trauma. 24 Xiong et al. 25 have indicated that the deactivation of the p38 MAPK pathway can alleviate the severity of intestinal ischemia-reperfusion–induced ALI. 25 Thus, we wanted to know whether EPO can mediate the p38 MAPK pathway to affect I/R-induced ALI.
Hence, our study established the model of I/R-induced ALI in vivo and in vitro and determined the effect of EPO on ALI. In addition, we also detected the interaction of EPO with the p38 MAPK pathway in ALI induced by I/R.
Methods
Animals
A total of 48 Sprague Dawley rats (male, 250–300 g, 7–9 weeks) were purchased from Laboratory Animal Resources, Chinese Academy of Sciences (Beijing, China). The rats were housed separately in specific pathogen-free cages with relative humidity of 65% ± 10% at room temperature (25°C ± 3°C) and were allowed to access to food and water freely. The light/dark cycle in the cages alternated 12/12 h. Animal experiments were carried out based on the Guide for the Care and Use of Laboratory Animals and got approval from the Animal Ethical and Welfare Committee of Nanjing Medicine University.
Isolation and perfusion of lungs
First, our laboratory constructed isolated perfused rat lung as previously described.26,27 In brief, the rats were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg). The rats were ventilated with room air at 60 breaths/min at a tidal volume of 3 mL with a positive end-expiratory pressure of 1 cm H2O. After a median sternotomy was made, the right ventricle was injected with heparin (1 U/g of body weight). 10 mL of intracardiac blood from the right ventricle was withdrawn. An afferent cannula was passed through the pulmonary valve into the pulmonary artery. Then, a small incision was made in the left ventricle at the apex of the heart, and the mitral apparatus was dilated carefully. Subsequently, an effluent cannula was inserted into the left atrium through the mitral valve to collect the effluent perfusate for recirculation. Pressure transducers were used to monitor pulmonary arterial pressure and left atrial pressure (i.e., pulmonary venous pressure and PVP) from the side arm of the cannula. A roller pump was utilized to perfuse lungs using recirculated perfusate and maintained at a constant flow of 7 mL/min. The recirculated perfusate includes the above-collected blood and Krebs–Henseleit buffer (Sigma-Aldrich) containing 4% bovine serum albumin and 0.1% glucose. The weight changes of the isolated lung in situ were recorded in real-time using an electronic balance.
Study design
The rats were randomly divided into four groups: sham operation group (sham), EPO treatment + sham group (EPO), I/R-induced ALI group (I/R), and I/R + EPO group. Each group has 12 rats. After ventilation, the lungs in all groups were perfused for 15 min to equilibrate on the apparatus. In the I/R group, to initiate the ischemic period, the lungs were stopped from room air ventilation and perfusion and next maintained in the deflated state for 40 min. Saline solution (2 mL) was added to the perfusate after ischemia. Then, perfusion and ventilation were resumed to initiate the reperfusion period for 60 min. Hence, the lung I/R model had been established. In the I/R + EPO group, EPO (3000 U/kg) diluted in 2 mL saline solution was administered via the perfusate after ischemia. In the sham group, the rats had a thoracotomy, and their lungs were perfused for 100 min without ischemia after the stabilization period. Besides, saline solution (2 mL) was administered in these rats via the perfusate in the last hour of perfusion. In the EPO group, EPO (3000 U/kg) diluted in 2 mL saline solution was administered in sham rats via the perfusate in the last hour of perfusion.
Microvascular permeability
The pulmonary capillary filtration coefficient (Kf), an index of microvascular permeability to water, was detected from the change in lung weight induced by the elevation of venous pressure. During ventilation and lung perfusion, PVP was quickly enhanced by 10 H2O for at least 7 min. The slow, steady phase of weight gain as a function of time (△W/△T) was drawn on a semilogarithmic paper. Next, the slow component was extrapolated to zero time for estimation of the transcapillary filtration. Kf was defined as the y-intercept of the plot (in g·min−1) divided by the PVP (10 cm H2O) and lung weight, and was presented in whole units of g·min−1 cm H2O−1 × 100 g wet lung weight.
Protein concentration and cytokine levels in bronchoalveolar lavage fluid
After the above experiments, 2.5 mL of saline was used to lavage the left lung to obtain bronchoalveolar lavage fluid (BALF). Bronchoalveolar lavage fluid was centrifuged at room temperature for 10 min at 200 × g. After collecting the supernatant, the protein concentration in BALF was evaluated by a bicinchoninic acid (BCA) protein assay. Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) levels in BALF were measured by commercially available ELISA kits (R&D Systems Inc., USA).
Lung wet/dry weight ratio
One of indicators of pulmonary edema was lung wet/dry weight ratio. From each rat, the lower lobe of the right lung was collected and weighed. Then, the lungs were placed in a vacuum oven at 60°C for 48 h to achieve the dry weight. The wet/dry weight ratio was assessed.
Lung injury score
The lung tissues were fixed with 10% formaldehyde for 24 h and embedded in paraffin wax, followed by cutting into 2-μm–thick sections. Hematoxylin–eosin was used to stain the sections. A minimum of five randomly selected fields of each group were picked by two observers blinded to the study. The extent of lung injury was classified into a four-point scale (0) none, (1) mild, (2) moderate, or (3) severe and was scored based on presence or absence of interstitial infiltrate, alveolar integrity, and intra-alveolar fibrin.
Myeloperoxidase activity
The myeloperoxidase activity assay of lung tissues that is an indicator of neutrophil accumulation was performed in this study. Lung tissues were weighed and suspended in a hexadecyltrimethylammonium bromide buffer supplemented with potassium phosphate (50 mmol/l, PH 6.0). The tissues were homogenized at 5000 r/min in an ice bath (15 s/each time, four times), then frozen at −20°C, and subsequently thawed at room temperature four times. Next, the homogenate was centrifuged for 15 min at 30,000 × g at 4°C. 0.05 mL of the supernatant was added to 1.45 mL of a 50 m
Biochemical analysis
The levels of malondialdehyde (MDA) and the activity of superoxide dismutase (SOD) were used to indicate the severity of lung peroxidation injury. The frozen lung tissues were weighted and homogenized in a 0.1
Western blot
Total protein from lung tissues and BEAS-2B cells was extracted using an ice-cold lysis buffer and quantified by a BCA protein assay. Lung homogenates (30 μg) were equally loaded into a 12% SDS-PAGE gel. Then, the protein was transferred from the gel to the PVDF membrane. Next, the membrane was blocked for 1 h at room temperature using 5% nonfat milk and was incubated with the primary antibody at 4°C overnight. After three washes with TBST (5 min/each time), the membrane was incubated with the conjugated secondary antibodies at room temperature for 1 h. Subsequently, the membrane was washed with TBST three times and visualized with chemiluminescence reagents. The quantification analysis of the blot was assessed by ImageJ software. The primary antibodies used in this study are listed below: anti-Bcl-2 antibody (ab182858, dilution 1/2000), anti-cleaved caspase-3 antibody (ab32042, dilution 1/500), and anti-caspase-3 antibody (ab13847, dilution 1/500) were obtained from ABCAM; anti-phospho-p38 MAPK antibody (p-p38, #4511, dilution 1/1000), anti-p38 MAPK antibody (p38, #8690, dilution 1/1000), anti-ERK1/2 (#9102, dilution 1/1000), anti-phosho-ERK1/2 (#8544, dilution 1/1000), anti-JNK (#9252, dilution 1/1000), anti-phospho-JNK (#4668, dilution 1/1000), and anti-β-actin antibody (#4970, dilution 1/1000) were purchased from Cell Signaling Technology. The secondary antibodies (goat anti-rabbit IgG H&L (HRP), dilution 1/2000, ab205718) were purchased from ABCAM. β-actin in this assay was utilized as the internal control.
Induction of hypoxia-reoxygenation (H/R) in BEAS-2B cells
Human normal lung epithelial cells (BEAS-2B) were purchased from the Cell Bank, Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM (Dulbecco’s modified Eagle’s medium) containing 10% FBS (fetal bovine serum), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 humidified incubator.
To simulate hypothermic ischemia, BEAS-2B cells were placed in a hypoxia chamber (Billups-Rothenberg, CA) for 24 h at 37°C in fresh DMEM supplemented with or without EPO. After hypoxia, the cells were reoxygenated at normoxic conditions (21% O2) for 4 h. BEAS-2B cells in the control group were maintained under normoxic conditions in a fresh DMEM medium for 28 h at 37°C, and the EPO group was incubated under normoxic conditions in a fresh DMEM medium supplemented with EPO for 28 h at 37°C.
Cell viability assay
After H/R treatment, 10 μl of CCK-8 (Cell Counting kit-8) solution was added to the cells, followed by incubating for 1 h at 37°C. Subsequently, the absorbance of the sample at 450 nm was determined by a microplate reader.
Flow cytometry
After H/R treatment, BEAS-2B cells were harvested and washed with PBS, followed by centrifuging at 670 × g for 5 min. Next, the cells (1 × 106 cells/mL) were resuspended in 1× binding buffer solution (100 μL), and then added with propidium iodide (PI, 5 μl) and annexin V-FITC (5 μl). The samples were incubated for 15 min at room temperature in the dark. Next, the flow cytometer (Beckman, USA) was utilized to assess the apoptotic rate.
Reactive oxygen species production
After H/R treatment, BEAS-2B cells were cultured with DCFH-DA (10 μ
MDA and Glutathione activities in H/R treated-BEAS-2B cells
After H/R treatment, BEAS-2B cells were collected. The MDA and GSH activities of the cells were measured by the corresponding commercial kits including the MDA assay kit (Cayman Chemical Co.) and a GSH assay kit (Sigma) based on the manufacturer’s protocol.
Statistical analysis
All data from three or more independent experiments are expressed as means ± standard deviation (SD). The difference between two groups was assessed by the Student’s t-test, and the difference among multiple groups was analyzed using the ANOVA test by GraphPad Prism 7.04 software. A p-value < 0.05 was considered statistically significant.
Results
EPO attenuates I/R-induced lung injury
Compared with the sham group, the W/D ratio of the lungs in the I/R group was significantly increased (Figure 1(a)). Besides, the inflammatory factors (TNF-α and IL-6) in I/R-treated rats exhibited higher levels than those in the sham group (Figure 1(b)). Meanwhile, I/R treatment significantly induced the lung injury in comparison with the sham group (Figure 1(c)). As compared to the sham group, BALF protein content and Kf were considerably elevated in the I/R group (Figures 1(d) and (e)). Hence, these results indicated that the I/R-induced lung injury rat model had been successfully established. Additionally, the levels of each indicator in Figure 1 had no significant difference between EPO and sham groups, indicating EPO had no side effect on the rat lung. Furthermore, I/R increased the weight of lung, the release of inflammatory factors, the injury severity of lungs, BALF protein content, and Kf, while EPO treatment obviously attenuated these effects (Figure 1). Therefore, EPO attenuated I/R-induced lung injury. Erythropoietin attenuates I/R-induced lung injury (A) The image of the lung wet/dry weight (W/D) ratio indicating pulmonary edema. (B) The inflammatory factors (TNF-α and IL-6) in lung tissues were detected by commercially available ELISA kits. (C) Lung tissues were stained by hematoxylin–eosin to show the morphologic changes, and the bar graph shows the score of lung injury. (D) Protein concentration in bronchoalveolar lavage fluid was assessed by the bicinchoninic acid protein assay (E) The pulmonary capillary filtration coefficient (Kf) determined before (baseline) and after ischemia-reperfusion injury. **p < .01 versus the sham group; #p < .05, ##p < .01 versus the I/R group.
EPO inhibits apoptosis and oxidative damage induced by I/R
To detect the effect of EPO on I/R-induced apoptosis, the levels of antiapoptotic protein (Bcl-2) and apoptotic proteins (caspase 3 and cleaved caspase 3) were determined by Western blot. The decreased Bcl-2 and the increased cleaved caspase 3 induced by I/R were reversed by EPO treatment (Figure 2(a)). In addition, our study determined an indicator of the severity of lung peroxidation injury (MDA), an indicator of neutrophil accumulation (MPO) and an antioxidant enzyme (SOD) to indicate the effect of EPO on oxidative damage induced by I/R. Ischemia-reperfusion obviously upregulated the levels of MDA and the activity of MPO, but EPO treatment alleviated these effects (Figure 2(b)). The activity of SOD was suppressed by I/R, while its activity was restored by EPO (Figure 2(b)). Hence, EPO could inhibit apoptosis and ROS induced by I/R. Erythropoietin inhibits apoptosis and oxidative damage induced by I/R (A) The expression of antiapoptotic protein (Bcl-2) and apoptotic proteins (cleaved caspase 3 and caspase 3) were determined by Western blot. (B) The level of MDA and the activity of MPO and SOD were detected by corresponding commercial assay kits. **p < .01 versus sham group; ##p < .01 versus I/R group. Notes: MPO: myeloperoxidase activity; SOD: superoxide dismutase; I/R: ischemia-reperfusion; MDA: malondialdehyde.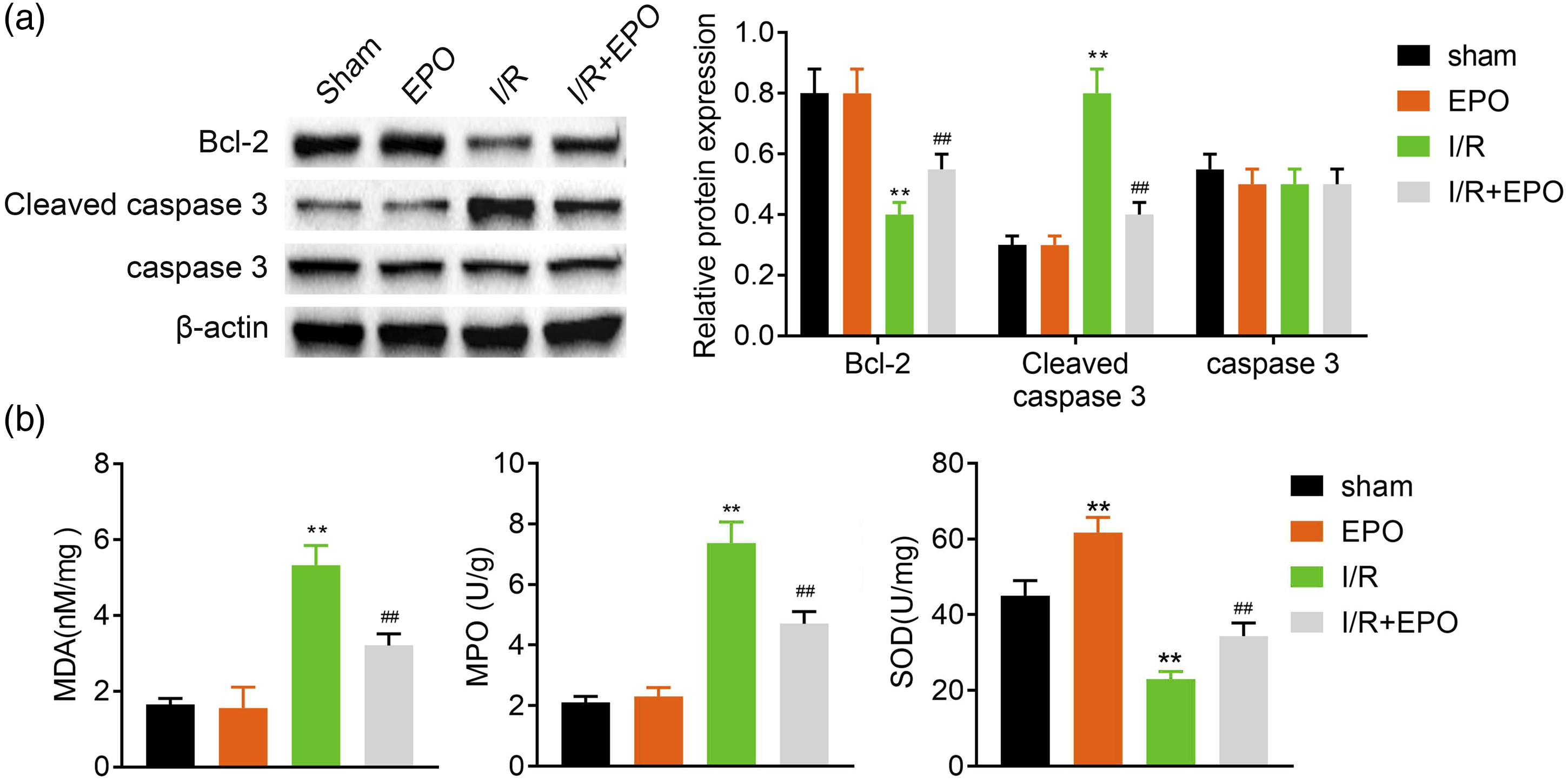
EPO suppresses the activation of the p38 MAPK signaling pathway caused by I/R
As shown in Figure 3, Western blot results indicated that I/R greatly increased the protein expression of p-p38, p-JNK, and p-ERK1/2 compared with the sham group, while the introduction of EPO attenuated the molecular expression. Thus, EPO suppressed the activation of the p38 signaling pathway caused by I/R. Erythropoietin suppresses the activation of p38 MAPK signaling caused by I/R. The expression levels of p38 MAPK (p38), p-p38 MAPK (p-p38), p-JNK, JNK, p-ERK1/2, and ERK1/2 in the p38 signaling pathway in lung tissues were measured by Western blot. **p < .01 versus the sham group; ##p < .01 versus the I/R group.
EPO attenuates the injury of BEAS-2B cells induced by H/R through blocking p38 signaling
To further investigate the protective mechanism of EPO in I/R-induced ALI, we constructed an I/R cell model by H/R treatment. The CCK-8 assay revealed that H/R impaired the viability of BEAS-2B cells, but EPO improved the situation (Figure 4(a)). The increased apoptosis induced by H/R was downregulated by the introduction of EPO (Figures 4(b) and (c)). H/R significantly enhanced the production of ROS, which was reversed by EPO treatment (Figure 4(d)). H/R caused the increase in MDA and the decrease in antioxidant GSH, and these changes were reversed by EPO treatment (Figure 4(e)). Moreover, H/R obviously upregulated the expression of p-p38, p-JNK, and p-ERK1/2, indicating the significant activation of the p38 signaling pathway (Figure 4(f)). The introduction of EPO attenuated the levels of p-p38, p-JNK, and p-ERK1/2 (Figure 4(f)). Collectively, EPO could attenuate H/R-induced injury of BEAS-2B cells through blocking p38 signaling. Erythropoietin attenuates the injury of BEAS-2B cells induced by H/R by blocking p38 signaling (A) The viability of BEAS-2B cells was detected by the CCK-8 assay. (B, C) The cellular apoptosis was determined using flow cytometry (D) The production of ROS was examined by a fluorescence microplate reader. (E) MDA and GSH activities of the cells were measured by the corresponding commercial kits (F) The expression levels of p38 MAPK (p38), p-p38 MAPK (p-p38), p-JNK, JNK, p-ERK1/2, and ERK1/2 in the p38 signaling pathway in BEAS-2B cells were measured by Western blot. *p < .05, **p < .01 versus the control group; #p < .05, ##p < .01 versus the H/R group. Notes: MDA: malondialdehyde; GSH: glutathione activities in H/R.
The inhibitor of p38 attenuates the injury of BEAS-2B cells induced by H/R
To validate the interaction between EPO and p38 signaling in H/R-induced lung cells, SB203580, as an inhibitor of p38,
28
was introduced in this study. Compared with the H/R group, both SB203580 and EPO could enhance the activity of BEAS-2B cells (Figure 5(a)), decrease the cellular apoptosis (Figures 5(b) and (c)), downregulate the levels of ROS and MDA (Figures 5(d) and (e)), and upregulate the levels of GSH (Figure 5(e)). SB203580 and EPO together further enhanced the activity of BEAS-2B cells (Figure 5(a)), reduced apoptosis (Figures 5(b) and (c)), and downregulated the levels of ROS and MDA compared with EPO only (Figures 5(d) and (e)). Thus, EPO along with SB203580 had more obvious protection effects than EPO alone. These findings indicated that SB203580 attenuated the injury of BEAS-2B cells induced by H/R, and further confirmed that EPO could regulate the activity of p38 signaling to improve the cellular injury induced by H/R. The inhibitor of p38 MAPK attenuates the injury of BEAS-2B cells induced by H/R (A) The viability of BEAS-2B cells was detected by CCK-8 assay. (B), (C) The cellular apoptosis was determined using flow cytometry (D) The production of ROS was examined by a fluorescence microplate reader. (E) MDA and GSH activities of the cells were measured by the corresponding commercial kits. *p < .05, **p < .01 versus the H/R group; #p < .05, ##p < .01 versus the H/R + EPO group. Notes: EPO: erythropoietin; MDA: malondialdehyde; GSH: glutathione activities in H/R; I/R: ischemia-reperfusion.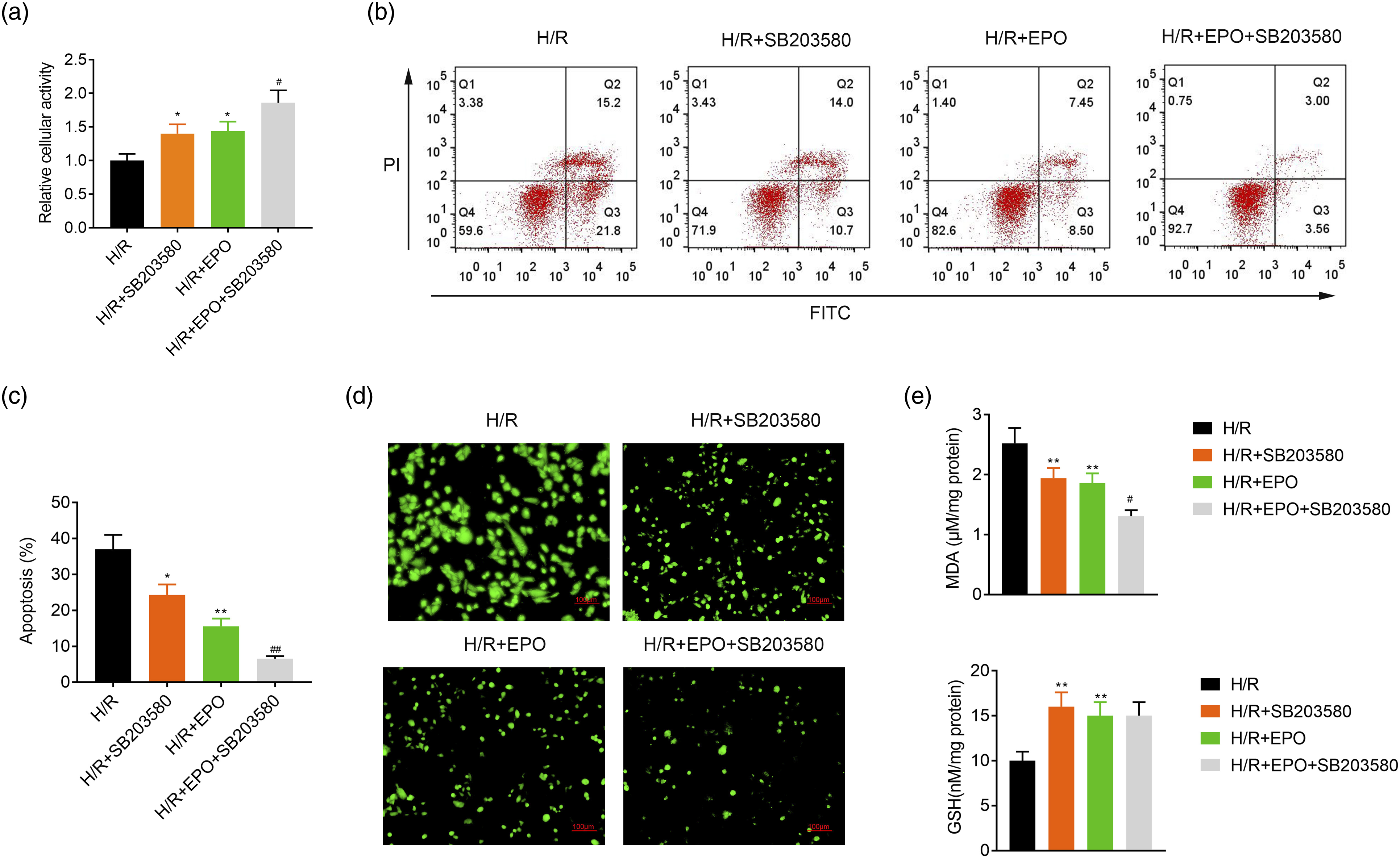
Discussion
It has been reported that I/R injury occurs in many clinical conditions such as lung transplantation and leads to significant morbidity as well as mortality. 29 The process of I/R injury is accompanied by innate immune responses, oxidative stress, cellular damage, and lung inflammation,3,30 but its specific molecular mechanisms have not been fully studied. Therefore, understanding the process of I/R injury is very important for finding new therapeutic options to reduce tissue injury. Hence, our study constructed the animal and cell models of I/R-induced lung injury to prepare the related research.
Erythropoietin, a glycoprotein cytokine, has a range of functions in addition to stimulation of erythropoiesis, including antiapoptotic, antioxidative, and anti-inflammatory effects on many diseases.6,8–10 For instance, EPO has been proved to reduce oxidative stress, suppress apoptosis, and decrease the release of inflammatory factors in lung injury induced by various risk factors.14–17 EPO reduces inflammasome and oxidative damage to ameliorate I/R-induced acute kidney injury. 31 Consistent with the above previous reports, our study showed that I/R induced the elevation of lung weight, inflammatory factors (TNF-α and IL-6) release, the lung injury severity, BALF protein content, and Kf, and the increase of these factors was significantly attenuated by EPO. Additionally, I/R increased apoptosis, MDA levels, and MPO activity but decreased SOD activity, and these changes were reversed by the introduction of EPO. Thus, EPO could attenuate the lung injury induced by I/R in vivo. Furthermore, H/R suppressed the viability of BEAS-2B cells, induced apoptosis, increased the levels of ROS and MDA, and decreased GSH levels, which were changed by EPO. Hence, EPO improved H/R-induced injury in vitro. Collectively, the protective effect of EPO was confirmed in I/R-induced lung injury in vivo and in vitro in this study. However, the detailed cellular and molecular signaling pathway of EPO needs further investigation.
p38 MAPK (p38) responds to various stress stimuli and is associated with cell autophagy, apoptosis, differentiation, and proliferation.19,20 Accumulating evidence indicate that the activation of the p38 MAPK signaling pathway aggravates lung injuries induced by I/R and is positively correlated with inflammatory factors (e.g., IL-1β and TNF-α) levels.32,33 Our study also showed that I/R-induced lung injury in vivo and in vitro significantly upregulated the expression of p-p38 MAPK, p-JNK, and p-ERK1/2 in comparison with the control group. Furthermore, EPO inhibited the expression of p-p38 MAPK, p-JNK, and p-ERK1/2, thus suppressing the activity of p38 MAPK signaling. Xing et al. show that the deactivation of the p38 MAPK signaling caused by epicatechin can alleviate inflammation in lipopolysaccharide-induced ALI. 34 Li et al. reveal that blocking p38 MAPK signaling suppresses the severity of ALI and the lung inflammation in vivo. 35 Hence, we speculated that EPO could block the p38 MAPK signaling pathway to alleviate the lung injury induced by I/R in vivo and in vitro. To validate this speculation, the inhibitor of p38 MAPK, SB203580, was introduced to treat the injury of BEAS-2B cells induced by H/R. The results indicated that both SB203580 and EPO promoted cell growth, inhibited apoptosis, downregulated the levels of ROS and MDA, and upregulated the levels of GSH by blocking p38 MAPK signaling. Moreover, EPO along with SB203580 had more obvious protection effects than EPO alone. Taken together, EPO alleviated the lung injury induced by I/R in vivo and in vitro by blocking p38 MAPK signaling.
In conclusion, EPO could inhibit the activity of p38 MAPK signaling pathway to attenuate I/R-induced ALI. The interaction mechanism of EPO with p38 MAPK signaling contributes to understanding the processes of I/R-induced ALI and provides new insights for the treatment of ALI induced by I/R. The toxicology, pharmacodynamics, and detailed mechanism of the protective effect of EPO on I/R-induced ALI remain to be elucidated by biological experiments and clinical trials in the future.
Footnotes
Acknowledgments
Not applicable.
Authors' contributions
Wei Zhao and Wei Gao designed the study and supervised the data collection, Ling Jia and Wenjing Cui analyzed the data and interpreted the data. Jiao Chen, Jinghui Yang, Xiang Xue, and Jianqin Cai prepared the article for publication and reviewed the draft of the article. All authors have read and approved the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the Municipal Science and Technology Plan (Guidance) Project of Nantong City in 2014 (Grant No. HS149147). Science and Technology Development Program of Nanjing Medical University (NMUB2019075), National Natural Science Foundation of China (81700331 and 81970217), Jiangsu Province Health Development Project with Science and Education (QNRC201685), Six One Project of Jiangsu Province (LGY2018100) and the Six Talent Peaks Project of Jiangsu Province (WSN-175).
Ethics approval
Animal experiments were carried out based on the Guide for the Care and Use of Laboratory Animals and got approval from the Animal Ethical and Welfare Committee of Nanjing Medicine University.
Statement of Informed Consent
Not applicable
Availability of data and materials
All data generated or analyzed during this study are included in this published article.