
Abstract
This study is aimed to investigate the role of long non-coding RNA 630 (LINC00630) in hepatocellular carcinoma (HCC). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed to examine LINC00630 expression in HCC cell lines and tissues. After LINC00630 was overexpressed or depleted in HCC cell lines, cell counting kit-8 (CCK-8) assay, BrdU assay, and flow cytometry were conducted for detecting HCC cell multiplication, apoptosis, and cell cycle progression. The catRAPID database was adopted to predict the binding relationship between LINC00630 and E2F transcription factor 1 (E2F1), and RNA pull-down and RNA immunoprecipitation (RIP) assays were carried out to verify this binding relationship. The binding of E2F1 to the cyclin-dependent kinase 2 (CDK2) promoter region was verified by dual-luciferase reporter gene assay and chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) assay. Western blotting was conducted to detect the protein expression of E2F1 and CDK2 in HCC cells. We report that LINC00630 expression was up-regulated in HCC and was significantly correlated with TNM stage and lymph node metastasis. LINC00630 overexpression facilitated HCC cell proliferation and cell cycle progression and inhibited the cell apoptosis, while LINC00630 knockdown had the opposite effects. LINC00630 directly bounds with E2F1. LINC00630 overexpression enhanced the binding of E2F1 to the CDK2 promoter region, thereby promoting CDK2 transcription, whereas knocking down LINC00630 inhibited CDK2 transcription. Collectively, LINC00630 promoted CDK2 transcription by recruiting E2F1 to the promoter region of CDK2, thereby promoting the malignant progression of HCC. Our data suggest that LINC00630 is a promising molecular target for HCC.
Keywords
Introduction
Known as the main pathological subtype of liver cancer, with about 800,000 new cases diagnosed annually, hepatocellular carcinoma (HCC) is one of the most common and aggressive malignancies.1,2 There have emerged studies showing that hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol abuse, liver cirrhosis, and smoking are risk factors of HCC.3,4 At present, the treatment options for HCC mainly include surgery, local ablation therapy, liver transplantation, molecular targeted therapy, systemic chemotherapy, etc. Despite steady progress in HCC diagnosis and treatment, the 5-year survival rate of HCC patients after surgical treatment is still low due to recurrence and metastasis.5,6 In this context, it is of great importance to clarify the mechanism of HCC tumorigenesis and seek for valuable biomarkers and novel therapeutic targets.
Long non-coding RNAs (lncRNAs) are a type of non-coding RNA with more than 200 bases in length . 7 LncRNA is closely related to cell survival, proliferation, differentiation, and migration. 8 LncRNAs feature prominently in regulating gene expression; their functions have been confirmed to be correlated with the tumorigenesis, development, and metastasis of various tumors.9-12 Previous studies have suggested that LINC00630 can facilitate non-small cell lung carcinoma (NSCLC) and colorectal carcinoma progression.13,14 Nevertheless, the biological functions and mechanism of LINC00630 in HCC are yet to be elucidated.
E2F transcription factor 1 (E2F1) belongs to the E2F family of transcription factors; it can take part in the regulation of cell proliferation, differentiation, and apoptosis and plays a tumor-promoting role in HCC.15,16 Cyclin-dependent kinase 2 (CDK2) belongs to the serine/threonine kinase family and mainly takes part in regulating the cell cycle; its high expression is associated with the development of multiple cancers including HCC.17,18 We report that LINC00630 is up-regulated in HCC, and its overexpression promotes the binding of the transcription factor E2F1 to the CDK2 promoter region to facilitate CDK2 transcription, thus promoting HCC cell multiplication, suppressing the apoptosis, and accelerating cell cycle progression. These findings reveal the biological functions of LINC00630 and suggest that LINC00630 may have the potential to be a HCC treatment target.
Materials and methods
Tissue collection
With the approval of the Ethics Review Board of Renmin Hospital of Wuhan University and the informed consent of all patients (approval number: 2016-08), we selected 63 patients with HCC who had previously been admitted to our hospital, and collected the cancerous tissues and the corresponding para-cancerous tissues after surgical resection. The surgically removed tissues were preserved in a −80°C refrigerator until RNA extraction. The clinical and pathological data of all subjects were complete, and none of them had undergone any neoadjuvant therapy such as radiotherapy or chemotherapy before operation.
Cell culture
HCC cell lines (Bel-7402, SK-Hep1, MHCC-97H, and HepG2) and normal liver cell line (L02) used in the present study were bought from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Cat# 12100-046, Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Shanghai, China), 100 U/ml penicillin, and 0.1 mg/mL streptomycin (Thermo Fisher Scientific, Shanghai, China) at 37°C in 5% CO2. When the cells grew to the logarithmic growth phase, they were harvested for subsequent experiments.
Cell transfection
Bel-7402 and HepG2 cells were seeded into 60-mm culture dishes (1 × 106 cells/ml), which were then cultured in 5% CO2 at 37°C. Cell transfection was conducted after 24 h. The pcDNA3.1 empty vector (oe-NC), LINC00630 overexpression pcDNA3.1 plasmid (oe-LINC00630, full-length LINC00630 was cloned into pcDNA3.1: NR_038988.2), small interfering RNA against LINC00630 (si-LINC00630-1: CATTCTCATTCAAGGGTCAATACTT; si-LINC00630-2: CAAGGGTCAATACTTAGGAGTTCAA), siRNAs against E2F1 (si-E2F1: GGACCACCTGATGAATATCTGTACT), negative control (si-NC: CATTACTAACTTGGGTAACACTCTT), and pcDNA-E2F1 overexpression plasmid (oe-E2F1, full-length LINC00630 was cloned into pcDNA3.1: NM_005225.3) were bought from RiboBio Co., Ltd. (Guangzhou, China). The transfection was conducted using Lipofectamine® 2000 kit (Cat# 11668019, Invitrogen, Carlsbad, CA, USA) by following the manufacturer’s instruction. After 24 h of transfection, Western blot and quantitative real-time polymerase chain reaction (qRT-PCR) were utilized for determining the transfection efficiency.
Quantitative real-time polymerase chain reaction
TRIzol reagent (Cat# 15596-018, Invitrogen, Carlsbad, CA, USA) was adopted for total RNA extraction from HCC tissues or cells. A Reverse Transcription Kit (Takara, Dalian, China) was used for the reverse transcription of RNA into cDNA. qRT-PCR was conducted using Power SYBR Green PCR Master Mix (Cat# 368706, Takara, Dalian, China) on ABI7300 PCR system (Cat# ABI-7300, Thermo Fisher Scientific, Shanghai, China). With GAPDH as the internal reference of LINC00630, the 2−ΔΔCt method was adopted to calculate the relative expression of LINC00630. LINC00630 primer sequence was as follows: forward, 5’-TACCGTTATTATTTCCC-3’; reverse, 5’-TCCTAAGTATTGACCCT-3’. GAPDH primer sequence was as follows: forward, 5’-ACAACTTTGGTATCGTGGAA-3’; reverse, 5’-GCCATCACGCCACAGTTTC-3’. U6 primer sequence was as follows: forward, 5’-CTCGCTTCGGCAGCACA-3’; reverse, 5’-AACGCTTCACGAATTTGCGT-3’.
Cell counting kit-8 assay
Cell viability was measured by a cell counting kit-8 (CCK8) detection kit (Dojindo Molecular Technologies, Kumamoto, Japan). The cells in each group were collected to prepare single-cell suspension. The cell density was adjusted after cell counting, and cells were transferred to 96-well plates with 1000 cells per well. The next day, after the cells adhered to the wall, 90 μL of medium and 10 μL of CCK-8 solution were added into each well. After incubation for 2 h, the optical density (OD) at 450 nm of each well was measured by a microplate reader and recorded. In this way, the OD values of the cells after 24, 48, 72, and 96 h of culture were determined. The cell growth curve was drawn with time as the x-coordinate and OD 450 nm as the y-coordinate.
BrdU assay
BrdU assay was performed using a BrdU labeling kit (Cat# 552598, BD Biosciences, San Jose, CA, USA). Bel-7402 and HepG2 cells were cultured on slides in 24-well plates. When the cells grew to the logarithmic growth phase, each well was added with 200 μL of 5 μmol/L BrdU working solution. The cells were incubated for 2 h in the cell incubator, and then the cells on the slides were washed with PBS and fixed with 2.5% paraformaldehyde at room temperature for 10 min. After the incubation with 200 μL of 2 mg/mL glycine for 5 min, each well was added with 100 μL of PBS containing 0.5% TritonX-100, and the incubator was put on the shaker for destaining for 10 min. After that, the cells were washed with PBS twice, 5 min for each time. Next, the cells were stained with Apollo in the dark for 30 min at room temperature, and then incubated with 1 × Hoechst 33342 DNA staining solution at room temperature in the dark for 20 min. After PBS washing, the cells were photographed and counted under the fluorescence microscope.
Flow cytometry
The AnnexinV-FITC/PI double staining kit (Cat# 556547, BD Biosciences, Bedford, MA, USA) was used for the detection of cell apoptosis. The cells during logarithmic growth were collected, and Bel-7402 and HepG2 cells were seeded at 8 × 103 cells/well into 96-well plates, which were subsequently incubated at 37°C in 5% CO2. After the cells adhered to the wall, the digested cells were centrifuged for 5 min at 2000 r/min, and the medium was discarded. The cells were rinsed with PBS and centrifuged for 5 min at 2000 r/min. After the supernatant was discarded, the cells were added with 5 μL of AnnexinV-FITC (BD Biosciences, Bedford, MA, USA). They were mixed gently and incubated for 15 min in the dark at room temperature. Subsequently, they were added with 10 μL of PI staining solution, mixed gently, and incubated in an ice bath for 5 min in the dark. Within 30 min, the flow cytometry detection was carried out. PI was for red fluorescence, and AnnexinV-FITC was for green fluorescence. The apoptosis index is calculated: [apoptosis index (%) = AnnexinV(+)PI(−)(%) + AnnexinV(+)PI(+)(%)].
The cell cycle was also analyzed by flow cytometry. Cells during logarithmic growth were transferred to 6-well plates. After 24 h of culturing, they were trypsinized, washed twice with PBS, fixed with 70% ethanol, and stored at 4°C overnight. Then, the cells were washed once with PBS, and the cell density was adjusted to 1 × 106 cells/ml with binding buffer. The PI staining solution was then added to reach a final concentration of 0.05 mg/mL, and the cells were then stained for 30 min at 4°C. The flow cytometer was used for analyzing the cell cycle.
Validation of Subcellular distribution of LINC00630
RNAs from the cytosol and nucleus of Bel-7402 and HepG2 cells were separated and collected with an NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Cat#78835, Thermo Fisher Scientific, Shanghai, China) and an RNeasy Kit (Cat# 74104, Qiagen, Shanghai, China), respectively. Then, qRT-PCR was conducted to measure LINC00630 expression in the cytosol and nucleus of HCC cells, respectively, with GAPDH and U6 as the controls.
RNA pull-down assay
Following the manufacturer’s protocol, the Magnetic RNA-Protein Pull-Down Kit (Cat# 2016420, Thermo Fisher Scientific, Shanghai, China) was employed for detecting the interaction between LINC00630 and E2F1. Protein extracts mixed with biotinylated LINC00630 were incubated with magnetic beads. Ultimately, the eluted proteins from the beads were utilized for Western blot to detect the enrichment of E2F1.
RNA immunoprecipitation (RIP) assay
Following the manufacturer’s instruction, the Magna RIP™ RNA binding protein immunoprecipitation kit (Cat# 17-700, Millipore, Billerica, MA, USA) was adopted to perform RIP assay. In short, HCC cell lysates were incubated with corresponding anti-E2F1 antibody or IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and the RNA–protein compounds were enriched by the magnetic beads, and then the RNA was purified, and the level of LINC00630 was detected by qRT-PCR. The total RNA acted as input control, and the antibody isotype IgG acted as negative control.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay was performed using the EZ-ChIPTM kit (Cat# 17-371, Millipore, Billerica, MA). The transfected Bel-7402 and HepG2 cells were cultured for 36 h, and subsequently fixed with 37% formalin. After incubation, the fixation was terminated with glycine, and the cells were scraped and centrifuged to obtain cell pellet. PMSF-containing cell lysis buffer was added to suspend the cells, which were then centrifuged, and the supernatant was removed to obtain the nuclear precipitates. After ultrasonic DNA shearing in an ice-water bath, lysates were divided into two tubes and incubated with antibody to IgG (ab172730, 1:100, Abcam, Shanghai, China) or antibody to E2F1 (KH-95, Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4°C. Subsequently, DNA–protein complex was precipitated using Protein Agarose, and the centrifugation was carried out at 12,000 g for 5 min at 4°C. Afterward, the cross-linking was reversed overnight at 65°C, and DNA fragments were collected, and purification was performed with phenol/chloroform. Last, qRT-PCR was conducted to detect the binding of E2F1 to the CDK2 promoter region with the CDK2-specific primer.
Dual-luciferase reporter gene assay
Dual-luciferase reporter assay system (Cat# 11752250, Promega, Madison, WI, USA) was utilized for performing dual-luciferase reporter gene assay. The target fragments of wild-type CDK2 and mutant CDK2 were constructed and integrated into the pGL3 vector (Cat# E1751, Promega, Madison, WI, USA) to construct pGL3–CDK2-wild type (CDK2-WT) and pGL3–CDK2-mutant (CDK2-MUT1, CDK2-MUT2, and CDK2-MUT1/2) reporter vectors (the target sequences were shown in supplementary material). Bel-7402 and HepG2 cells were co-transfected with CDK2-WT (or CDK2-MUT) and E2F1 overexpression plasmid (or oe-NC). After 48 h of transfection, following the manufacturer’s instruction, the luciferase activity was determined.
Western blot
Cells were lysed with RIPA lysis buffer (Cat# P0013B, Beyotime Biotechnology, Shanghai, China) containing protease inhibitors. The supernatant was collected after high-speed centrifugation and heated at 100°C in a water bath for 10 min to denature the proteins. After protein quantification by the BCA method, SDS-PAGE was performed, and then the proteins were transferred onto the PVDF membrane. Subsequently, the PVDF membranes were washed with Tris Buffered Saline with Tween 20 (TBST), and then they were incubated with rabbit anti-E2F1 antibody (Abcam, ab179445, 1:500), rabbit anti-CDK2 antibody (Abcam, ab32147, 1:500), and rabbit anti-GAPDH antibody (Abcam, ab9485, 1:500) overnight at 4°C, respectively. After the membranes were rinsed with TBST, the membranes and goat anti-rabbit IgG H&L (HRP) (Abcam, ab150077, 1:1000) were incubated at room temperature for 1 h. After the membranes were washed with TBST again, the hyper-sensitive ECL kit was used for developing the protein bands. All the antibodies used in this work were bought from Abcam (Shanghai, China). The relative expressions of the proteins were calculated with ImageJ (NIH, Bethesda, Maryland, USA), with GAPDH as the endogenous control.
Statistical analysis
All experiments were repeated three times, with three replicates. SPSS 20.0 statistical software (SPSS Inc., Chicago, IL, USA) was adopted for the statistical analysis of experimental results. Measurement data were expressed as “mean ± standard deviation”. Student’s t-test was used to perform the comparison between two groups, and the comparison of means of multiple groups was performed with one-way analysis of variance. Enumeration data were presented in contingency table, and χ2 test was employed for the analysis of the difference between two groups. When p < 0.05, the difference was statistically significant.
Results
LINC00630 expression is up-regulated in HCC
By analyzing the HCC-related data in the GEPIA database (http://gepia.cancer-pku.cn/), it was found that LINC00630 expression in HCC tissue samples was significantly up-regulated (Figure 1(a)). Subsequently, LINC00630 expression in HCC cells and tissues was detected by qRT-PCR, and it was also revealed that LINC00630 expression in HCC tissues and cells was significantly increased compared with adjacent tissues or normal liver cells (Figures 1(b) and (c)). The chi-square test indicated that high LINC00630 expression was associated with the advanced TNM stage and lymph node metastasis in HCC (Table 1). LINC00630 expression in HCC is up-regulated. (a) LINC00630 expression characteristics in HCC tissue samples were analyzed using the GEPIA database. (b) and (c) Detection by qRT-PCR of LINC00630 expression in HCC tissues and cells. *** p < 0.001. Correlation between clinicopathological features and LINC00630 expression in HCC tissues. *p < 0.05.

Regulatory effects of LINC00630 on HCC cell multiplication, apoptosis, and cycle
To further explore the biological functions of LINC00630 in HCC cells, LINC00630 overexpression plasmid, si-LINC00630-1, and si-LINC00630-2 were transfected into Bel-7402 and HepG2 cells, and the qRT-PCR results showed that the transfection was successful (Figure 2(a)). CCK-8 and BrdU assays showed that LINC00630 overexpression could markedly promote HCC cell proliferation, while LINC00630 knockdown inhibited the cell proliferation (Figures 2(b) and (c)). Then, flow cytometry was utilized for detecting cell apoptosis and cell cycle, and it was discovered that LINC00630 overexpression could significantly inhibit cell apoptosis and accelerate cell cycle progression, whereas knocking down LINC00630 promoted HCC cell apoptosis and induced cell cycle arrest at the G0/G1 phase (Figures 2(d) and (e)). Biological functions of LINC00630 in HCC cells. (a) Detection by qRT-PCR of the transfection efficiency of LINC00630 overexpression plasmid, si-LINC00630-1, and si-LINC00630-2. (b) and (c) CCK-8 and BrdU assays were conducted to detect the effects of LINC00630 overexpression and knockdown on HCC cell proliferation. (d) and (e) Flow cytometry was performed to detect the effects of LINC00630 overexpression and knockdown on HCC cell apoptosis and cell cycle.* p < 0.05, ** p < 0.01 and *** p < 0.001.
LINC00630 interacts with transcription factor E2F1
To further understand the mechanism of LNC00630 in HCC cells, the LncMAP database (http://bio-bigdata.hrbmu.edu.cn/LncMAP/) was used to perform bioinformatics analysis, and it was shown that LINC00630 could regulate CDK2 transduction by recruiting E2F1 to the CDK2 promoter region (Figure 3(a)). Next, qRT-PCR was carried out to detect the subcellular localization of LINC00630 in Bel-7402 and HepG2 cells, and it was unveiled that LINC00630 was mainly distributed in cell nucleus (Figure 3(b)). Subsequently, the catRAPID online analysis tool (http://service.tartaglialab.com/page/catrapid_group) was used to predict the position of the binding domain between LINC00630 and E2F1, and the interaction spectrum results showed that LINC00630 could directly interact with E2F1 (Figure 3(c)). To further verify their interaction, RNA pull-down and RIP assays were conducted. RNA pull-down assay manifested that E2F1 expression in the biotinylated LINC00630 group was markedly up-regulated (Figure 3(d)). RIP assay suggested that LINC00630 was remarkably enriched by anti-E2F1 antibody both in Bel-7402 and HepG2 cells (Figure 3(e)). These data verified that E2F1 and LINC00630 could interact with each other. The binding relationship between LINC00630 and E2F1. (a) LncMAP database was used to predict the binding relationship among LINC00630, E2F1, and CDK2. (b) The subcellular localization of LINC00630 was examined by qRT-PCR after the cytosol and nucleus of HCC cells were separated. (c) The catRAPID online analysis tool was utilized to predict the binding domain between LINC00630 and E2F1. (d) (e) RNA pull-down and RIP assays were conducted to analyze the binding relationship between LINC00630 and E2F1. *** p < 0.001.
LINC00630 promotes CDK2 transcription by recruiting the transcription factor E2F1 to the CDK2 promoter region
PROMO database (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo) implied that E2F1 could bind to the CDK2 promoter region (Figure 4(a)). Moreover, the StarBase database (http://starbase.sysu.edu.cn/) indicated that LINC00630 and E2F1 expressions were both positively correlated with CDK2 expression in HCC tissues (Figure 4(b)). To verify their interaction, the dual-luciferase reporter gene assay was conducted. The results indicated that E2F1 overexpression could notably elevate the luciferase activity of WT CDK2, while knocking down E2F1 markedly suppressed the luciferase activity of WT CDK2 (Figure 4(c)). Subsequently, the aforementioned binding sequences were truncated or mutated, and dual-luciferase reporter gene assay was performed again. The results showed that in contrast to the WT groups, after site one was truncated or mutated, the luciferase activity of the reporter was significantly inhibited (Figures 4(d) and (e)); after both sites were truncated or mutated, E2F1 overexpression had little effect on the luciferase activity (Figures 4(d) and (e)). Subsequently, the ChIP-qPCR assay was conducted using E2F1-specific antibodies. In comparison with the sample bounded with IgG, we found that E2F1-bound complex showed a remarkable enrichment of CDK2 promoter (Figure 4(f)). In addition, LINC00630 overexpression enhanced the binding between E2F1 and CDK2 in Bel-7402 and HepG2 cells (Figure 4(g)). Western blotting and qRT-PCR showed that LINC00630 overexpression promoted CDK2 expression, whereas E2F1 knockdown weakened this effect; LINC00630 knockdown inhibited CDK2 expression, while E2F1 overexpression restored this effect (Figure 4(h)). Effect of LINC00630 on CDK2 expression by binding to E2F1. (a) Prediction of the binding sites between E2F1 and CDK2 promoter region through the PROMO database. (b) The StarBase database analysis of the correlation among LINC00630, E2F1, and CDK2 expressions. (c) Dual-luciferase reporter gene assay was performed to detect the effects of E2F1 overexpression and knockdown on the CDK2 promoter activity in HCC cells. (d) and (e) Dual-luciferase reporter gene assay was conducted to verify the binding sites between E2F1 and CDK2 promoter region. (f) ChIP-qPCR assay was applied to detect the binding of E2F1 to the CDK2 promoter region. (g) ChIP-qPCR assay was conducted to analyze the effect of LINC00630 on the enrichment of E2F1 in the CDK2 promoter region. (h) Western blot and qRT-PCR were carried out to detect the effects of LINC00630 and E2F1 on CDK2 protein and mRNA expressions. * p < 0.05, ** p < 0.01 and *** p < 0.001.
Effects of LINC00630/E2F1 axis on HCC cell multiplication, apoptosis, and cycle
Subsequently, we further investigated the regulatory effects of LINC00630/E2F1 axis on HCC cell proliferation, apoptosis, and cell cycle. CCK-8 assay, BrdU assay, and flow cytometry manifested that LINC00630 overexpression could facilitate HCC cell multiplication, inhibit cell apoptosis, and accelerate cell cycle (Figures 5(a)-(d)), while E2F1 knockdown could weaken these effects; knocking down LINC00630 could restrain HCC cell multiplication and induce cell apoptosis and cell cycle arrest at the G0/G1 phase, whereas E2F1 knockdown could restore these effects (Figures 5(a)-(d)). These data further supported that the biological function of LINC00630 in HCC was partly dependent on E2F1. Effects of LINC00630 and E2F1 on HCC cell proliferation, apoptosis, and cell cycle. Control plasmids, LINC00630 overexpression plasmids, and LINC00630 overexpression plasmids + si-E2F1 were transfected into Bel-7402 cells, respectively. Control siRNA, si-LINC00662-2, and si-LINC00662 + E2F1 overexpression plasmids were transfected into HepG2 cells, respectively. (a) and (b) CCK-8 and BrdU assays were used to detect the effects of LINC00630 and E2F1 on HCC cell proliferation. (c) and (d) Flow cytometry was conducted to detect the effects of LINC00630 and E2F1 on HCC cell apoptosis and cell cycle. * p < 0.05, ** p < 0.01 and *** p < 0.001.
Discussion
Over the past few years, increasing lncRNAs have been discovered. They can be divided into intronic lncRNA, intergenic lncRNA, sense lncRNA, bidirectional lncRNA, and antisense lncRNA.19,20 Although lncRNAs do not encode protein themselves, they take part in the regulation of gene expression at the transcriptional and post-transcriptional levels, and they feature prominently in various biological processes such as dosage compensation effect, epigenetic regulation, cell cycle, transcriptional interference, and protein activation.21,22 In HCC, lncRNAs can act as carcinogenic or tumor-suppressing factors to participate in its occurrence and development. For example, lncRNA uc.134 can inhibit CUL4A-mediated LATS1 ubiquitination and increase YAPS127 phosphorylation, thereby silencing the target genes of YAP to inhibit HCC cell proliferation and metastasis in vivo. 23 LncRNA AY927503 (AY) is highly expressed in HCC and is associated with the patient’s poor prognosis; AY can specifically interact with the integrin αV (ITGAV) promoter to promote ITGAV transcription and αVβ3 expression, thereby facilitating the metastasis of HCC. 24 Furthermore, LINC00630 can facilitate the metastasis of NSCLC, via the DDX23-Linc00630-HDAC1 axis to activate the Notch signaling pathway; 13 LINC00630 and EZH2 negatively regulate BEX1 through the methylation of promoter DNA to promote colorectal cancer progression. 14 This study found that LINC00630 expression was enhanced in HCC tissues, and its high expression was associated with the advancing of TNM stage and lymph node metastasis of HCC; functional experiments showed that LINC00630 overexpression could promote HCC cell proliferation, inhibit cell apoptosis, and accelerate cell cycle progression, while LINC00630 knockdown had the opposite effects. These suggest that LINC00630 plays a cancer-promoting role in HCC.
The E2F family includes eight members, E2Fl–E2F8, and their corresponding genes are located on different chromosomes and carry a high degree of gene homology. 25 E2F1 can promote cell multiplication via accelerating cell cycle progression from G1 phase to S phase. 26 At present, a lot of studies report that E2F1 features prominently in HCC and is related to its occurrence and development. For example, down-regulating E2F1 expression leads to a decline in the expressions of cyclin A2, cyclin E1, and CDK2, and blocking E2F1 can suppress HCC cell proliferation and invasion; 15 E2F1 overexpression can activate cell cycle-related factors and the phosphorylation of PKCα to promote HCC cell proliferation. 27 It is noteworthy that the regulatory effect of E2F1 on downstream target gene transcription is also modulated by lncRNAs. For instance, lncRNA-HIT can interact with E2F1 to regulate the expression of its target genes Survivin, FOXM1, SKP2, NELL2, and DOK1, therefore potentiating the growth of NSCLC cells; 28 lncRNA NR-104098 can recruit E2F1 to the EZH2 promoter region by binding to E2F1 to inhibit EZH2 transcription, thereby inhibiting the progression of acute myeloid leukemia. 29 In this work, we demonstrated that LINC00630 could interact with E2F1, suggesting that LINC00630 is a novel regulator of E2F1’s transcriptional activity.
Cell cycle dysregulation features prominently in tumorigenesis, and CDK is one of the key molecules in regulating cell cycle; the excessive activation of CDKs results in premature DNA replication in cells, leading to genome instability and inducing tumorigenesis. 30 Additionally, CDK complexes regulate the phosphorylation of different substrate proteins, and thus to modulate the specific transcription of a series of downstream genes, therefore exerting their regulatory effects in different phases of cell cycle.31,32 CDK2 is a member of the CDK family and can regulate HCC progression by changing cell proliferation and apoptosis. For example, SMYD3 can bind to the CDK2 and MMP2 promoters to increase their expressions, thereby facilitating the tumorigenicity and intrahepatic metastasis of HCC; 33 HOXA7 can promote HCC cell proliferation by increasing cyclin E1 and CDK2 expressions. 34 The present study revealed that LINC00630 overexpression could promote the binding of E2F1 to the CDK2 promoter region and thus facilitate CDK2 expression, finally promoting HCC cell multiplication, suppressing cell apoptosis, and accelerating cell cycle, while knocking down LINC00630 had the opposite effects. Our work partly explained the mechanism of CDK2 dysregulation in HCC.
To sum up, for the first time, our study shows that LINC00630 expression is increased in HCC tissues and cells, and its high expression is associated with the poor clinicopathological indicators of HCC patients. Functionally and mechanistically, it is confirmed that LINC00630 promotes CDK2 transcription by recruiting E2F1 to the CDK2 promoter region, thereby promoting the malignant progression of HCC (Figure 6). Our work implies that LINC00630 has the potential to become a diagnostic marker and molecular therapy target for HCC. However, there are several limitations of this work. First, the mechanism of the abnormally high expression of LINC00630 is unclear; second, the in vivo models are needed to further verify our findings. Graphic Abstract: LINC00630 promotes HCC progression via regulating E2F1/CDK2.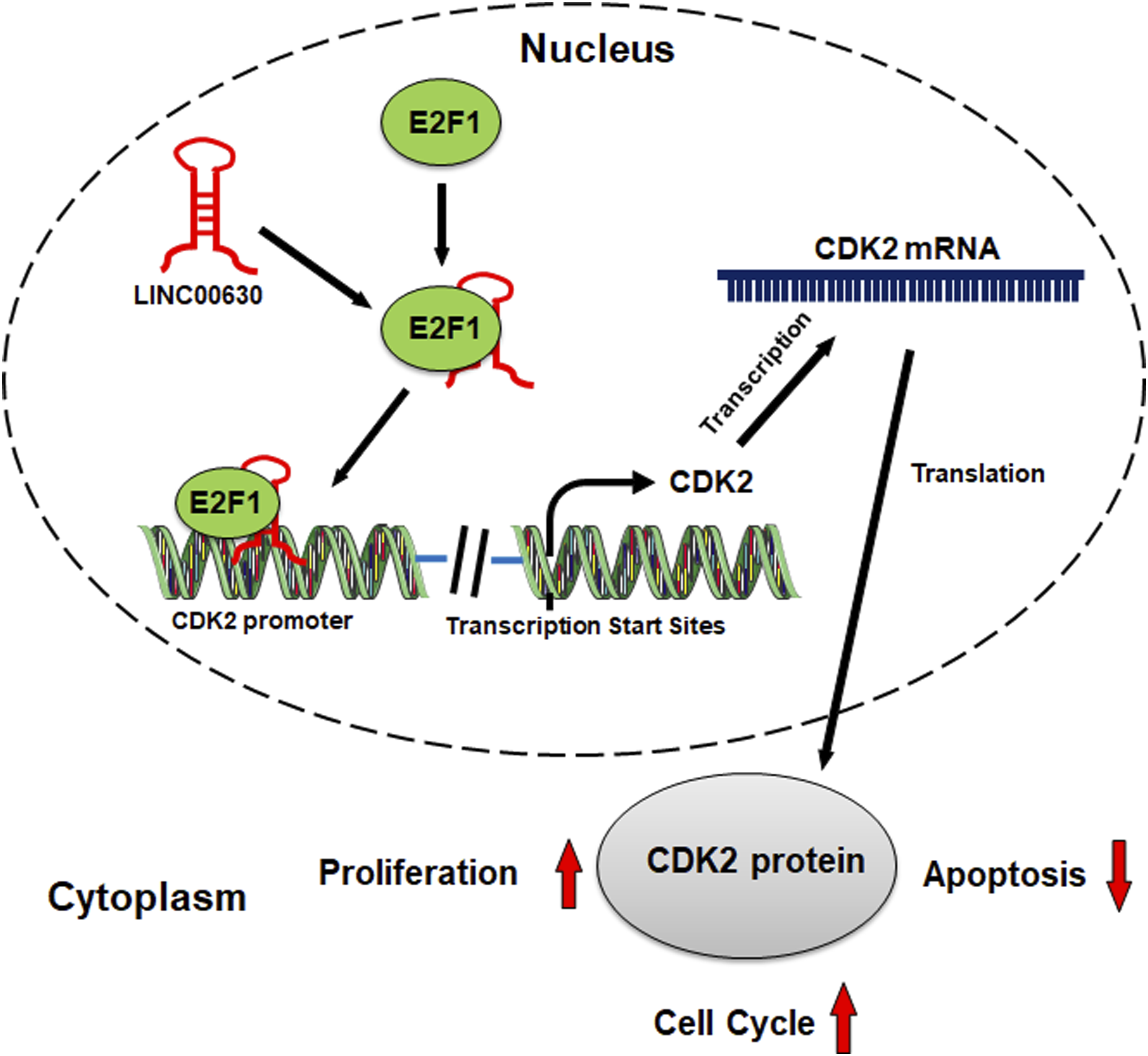
Footnotes
Acknowledgments
We thank Hubei Yican Health Industry Co., Ltd. for its linguistic assistance during the preparation of this manuscript.
Author’s Contribution
WGD and JK: Conceptualization and the design of the experiments. JK, XH, and WGD: Experiments conduction. JK, XYZ, and ML: Data curation and statistical analysis. JK, XH, ML, and NC: Original draft preparation. WGD and JK: Reviewing and revising manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
This study, including the collection of hepatocellular carcinoma tissue samples and adjacent tissue samples, was reviewed and approved by the Ethics Review Board of Renmin Hospital of Wuhan University. All protocols of experiments were in line with the Declaration of Helsinki.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.