
Abstract
The inflammation and immune responses are critical in ischemic stroke and contribute to aggravated brain damage. Ephedrine was reported to play an important role in the control of inflammatory responses. This study was to investigate the repairing effects and potential mechanisms of ephedrine on cerebral ischemic injury in a rat model of focal cerebral ischemia. The rat model of cerebral ischemia/reperfusion injury was established using the middle cerebral artery occlusion (MCAO) method and then rats were treated with ephedrine (5 and 10 mg/kg) for 7 days. The neurobehavioral progression was assessed using the neurological scoring method. The pathology of brain tissue was evaluated by hematoxylin and eosin (H&E) staining. The infarct volume was examined by triphenyltetrazolium chloride (TTC) staining. The apoptosis in ischemic brain tissues was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. Inflammatory factors were detected by enzyme-linked immunosorbent assay (ELISA). Gene quantification and protein expression were detected by real-time PCR and western blot, respectively. Ephedrine treatment significantly alleviated the cerebral ischemia/reperfusion injury, evidenced by decreased neurological deficit score, infarct volume and water content. Ephedrine also decreased autophagy and apoptosis in brain tissues. Moreover, ephedrine treatment significantly reduced inflammatory responses, associating with decreasing the protein expression of p-NF-κB. These results demonstrated neuroprotective properties of ephedrine and highlighted it as a new potential anti-inflammatory agent against injury of cerebral ischemia/reperfusion.
Keywords
Introduction
Although death from stroke has decreased over the past decade, with the fifth leading cause of death in the United States, the global burden of stroke is increasing. Stroke is now increasingly recognized as an important cause of cognitive problems and has been implicated in the development of both vascular dementia and Alzheimer’s disease. 1,2 There are two types of stroke, ischemic and hemorrhagic. 3 Ischemic stroke is the most prevalent cause and patients over 65 years old tend to remain in disability 6 months after stroke onset. Moreover, the cerebral ischemic injury is the leading cause of neurological function loss and neuronal death, and the exact mechanism has not been elucidated. 4 Several mechanisms are thought to be involved in the pathogenesis of ischemic stroke, including oxidative stress, excitatory amino acid toxicity, peroxidation, inflammation, free radical production, brain edema and neuronal apoptosis. 5,6
Post-ischemic inflammation induced by the immune response is an essential step in the progression of cerebral ischemia injury. 7,8 Inflammatory responses play a critical role in the pathogenesis of ischemic stroke, and the infiltration of various types of inflammatory cells to ischemic regions exacerbates ischemic injury. Following cerebral ischemia, inflammatory cytokines such as interleukin-1 beta (IL-1β), IL-6 and tumor necrosis factor-alpha (TNFα) increased. 9 –11 Several experimental models of stroke have demonstrated that inflammatory mediators contribute to the development of brain lesions and neurological deficits. 12 Evidence has been found that macrophages derived from glial cells and circulating monocytes also contribute to acute brain injury. 13 Peripheral white blood cells, including T cells, B cells and neutrophils, can aggravate brain ischemia in the early stages of stroke while releasing pro-inflammatory mediators. 14 Moreover, patients with metabolic syndrome who experience an ischemic stroke have higher levels of inflammatory markers. 15 All of these studies have shown that inflammatory responses play an important role in the tissue damage after cerebral ischemia.
In the treatment of ischemic stroke, inhibiting the process of ischemia/reperfusion and related pathological mechanisms is extremely important. Agents that can suppress specific damage mechanism are employed as therapeutic drugs. Ephedrine plays a classical role as an α- and β-adrenergic agonist, stimulating the central nervous system, elevating blood pressure and dilating bronchial tubes. 16 Ephedrine was reported to control the inflammatory responses by increasing IL-10 and decreasing pro-inflammatory cytokine (IL-1β, IL-6, IL-12 and TNFα) expression in primary peritoneal macrophages and Raw264.7 cells treated with peptidoglycan through phosphatidylinositol 3/Akt pathway. Moreover, the anti-inflammatory role of ephedrine was also demonstrated in an experimental mouse model of endotoxic shock by inhibiting pro-inflammatory cytokine secretion in response to lipopolysaccharide (LPS). 17,18 However, effects of ephedrine on inflammatory responses in ischemic stroke remains unclear. Therefore, this study was to investigate the neuroprotective effects of ephedrine in middle cerebral artery occlusion (MCAO) rat model and its underlying mechanisms.
Materials and methods
Chemicals and reagents
Caspase-3/9 activity assay kits and cytochrome-c assay kit were purchased from R&D systems (Minneapolis, MN, USA). TUNEL assay kit was purchased from Roche (Mannheim, Germany). IL-1β, IL-6 and TNFα assay kits were purchased from Thermo Fisher (Waltham, MA, USA). Ephedrine was purchased from National Institutes for Food and Drug Control (Beijing, China) with purity more than 99%. Ephedrine was dissolved in saline for treatment.
Experimental procedures and treatment
Male Sprague Dawley (SD) rats weighing 180–220 g were purchased from Charles River Ltd. (Beijing, China). Rats were maintained in humidity (40–70%) and temperature-controlled (23 ± 2°C) facility on 12:12 hour light:dark cycles under specific pathogen-free condition. Rats had free access to food and water during the experimental period. All animal procedures were approved by the Ethical Committee on Animal Care and Use of Yiwu Central Hospital (protocol No.: 2018-1518).
The Longa’s middle cerebral artery suture-occluded method was applied to prepare the cerebral ischemia/reperfusion (I/R) injury model. 19,20 Briefly, rats were fasted for 12 h before surgery with water accessible and then injected with 10% chloral hydrate (0.3 ml/100 g) intraperitoneally for anesthesia. A 2-cm incision was made in the middle of the neck. The right common carotid artery was exposed through a midline cervical incision. The right external carotid artery was dissected and isolated distally by coagulating its branches and placing a distal ligation prior to transection. A 3.0 surgical nylon suture was inserted into the lumen of the right external carotid artery stump and gently advanced into the internal carotid artery from the bifurcation. A sense of resistance indicated that the occlusion sutures reached the initial segment of the middle cerebral artery. Then the insertion of the occlusion sutures stopped and the surgical suture was tightly tied. After 2 h of ischemia, the sutures were pulled back to initiate a reperfusion process. Rats were kept in a thermal incubator to maintain their body temperature. The sham operation group was not subjected to ligation and suture insertion, but the rest procedures were the same.
The dosage selection of ephedrine was based on our preliminary study and the literature. 21 Two treatments of 20 mg/kg ephedrine demonstrated potent anti-inflammatory activity against D-GalN/LPS-induced acute liver failure in rats, through reducing the infiltration of inflammatory cells and lowering the production of inflammatory factors. 21 The experimental design was the following: Rat model of cerebral I/R injury was established by the MCAO method and rats showing abnormal post-operative condition were excluded from the study. Rats were randomly divided into four groups (n = 20): sham group, model group, ephedrine groups (5 or 10 mg/kg). Rats were treated at 30 min after ischemia. Rats in ephedrine groups received a daily dose of 5 or 10 mg/kg intraperitoneally, respectively, for 7 days. Rats in the sham group and model group received saline during the same period. 24 h after the last treatment, the neurobehavioral progression was assessed using the neurological scoring method. Brains were harvested to measure the change of pathology by H&E staining, triphenyltetrazolium chloride (TTC) staining, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. Inflammatory factors were detected by enzyme-linked immunosorbent assay (ELISA). Gene quantification and protein expression were detected by real-time PCR and western blot, respectively.
Neurological deficits
Neurological status was assessed blindly using a five-point scale (0–4) Bederson’s test by an investigator. 22
Triphenyltetrazolium chloride (TTC) staining
After treatment, rats were sacrificed and brains were quickly removed. Brains were cut into 2 mm-thick coronal sections. Sections were incubated with 2% TTC for 30 min at 37°C in the dark. The stained sections were scanned. The infarct volume was determined by image analysis and expressed as the percentage of the whole cerebral tissue.
Brain tissue water content measurement
After treatment, rats were sacrificed and brains were quickly removed. The wet weight (WW) was immediately weighed. Brains were then dried to obtain a dry weight (DW). Water content was calculated as the formula: (WW − DW)/WW × 100%.
Hematoxylin and eosin (H&E) staining
After treatment, rats were sacrificed and brains were quickly removed. Brains were fixed with 4% formalin for 24 h, and embedded in paraffin. Serial 5-µm sections of brain tissues were stained with H&E for microscopic examination. The histology was assessed by an experienced pathologist who was blind to experimental groups using an Olympus BX60 microscope (Olympus, Tokyo, Japan).
TUNEL assay
Apoptotic neuronal cells were detected by TUNEL staining according to the manufacturer’s instructions. Photographs were taken by an Olympus BX60 microscope (Olympus, Tokyo, Japan).
Cytochrome-c release measurement
Cytochrome-c in brain tissue lysate was measured by the assay kit according to the manufacturer’s instruction. After reaction with reagents, the optical density was measured at 450 nm by a microplate reader.
Caspase activity measurement
Caspase-3/9 activities in brain tissue lysate were measured by the assay kits according to the manufacturer’s instructions with the microplate reader.
Cytokine and nitric oxide (NO) measurement
Cytokine levels of IL-1β, IL-6 and TNFα in brain tissue lysate were measured by ELISA kits according to the manufacturer’s instructions. NO was measured by Griess assay (Sigma, St. Louis, USA).
Real-time PCR
Total RNA was extracted from brain tissue with the TaKaRa RNeasy reagents according to the manufacturer’s instructions. The RNA concentration was quantified by a spectrophotometer and mRNA was transcribed into cDNA with SuperScript master mix (Biorad). Quantitative PCR was run on StepOne systems using SYBR green Supermix (Thermo Fisher). The ΔΔCT method was used to quantify expression levels of target genes in different samples based on normalization to the level of β-actin.
Western blot analysis
The brain tissues were homogenized and lysed with RIPA buffer supplemented with protease and phosphatase inhibitor cocktails. Proteins (40 μg) were separated by 4–12% sodium dodecyl sulfate polyacrylamide gel and transferred to PVDF membranes. After blocking with 1% bovine serum albumin, the PVDF membranes were incubated with primary antibodies at 4°C overnight. The membranes were washed three times, followed by incubation with secondary antibody. The membranes were then washed three times, and proteins were visualized with an ECL detection reagents.
Statistical analysis
Data were expressed as mean ± standard deviation (SD) and analyzed with SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Spearman rank correlations were used to assess the association between the neurological deficit score and the percentage of infarct volume and water content. One-way analysis of variance was used for comparisons among multiple groups followed by LSD post hoc analysis. p < 0.05 was considered as significant difference.
Results
Ephedrine treatment ameliorated brain damage caused by I/R
After I/R, neurological deficit score of MCAO rats increased significantly, compared with sham group. However, ephedrine treatment significantly decreased the neurological deficit score. Moreover, at the end of treatment, brains were harvested for histopathological analysis. Ephedrine treatment obviously attenuated the damage in hippocampus caused by I/R. In addition, the percentage of infarct volume and water content was significantly diminished after ephedrine treatment, indicating the brain damage was ameliorated (Figure 1). The neurological deficit score was in good correlation with cerebral infarct size and water content (p < 0.05).
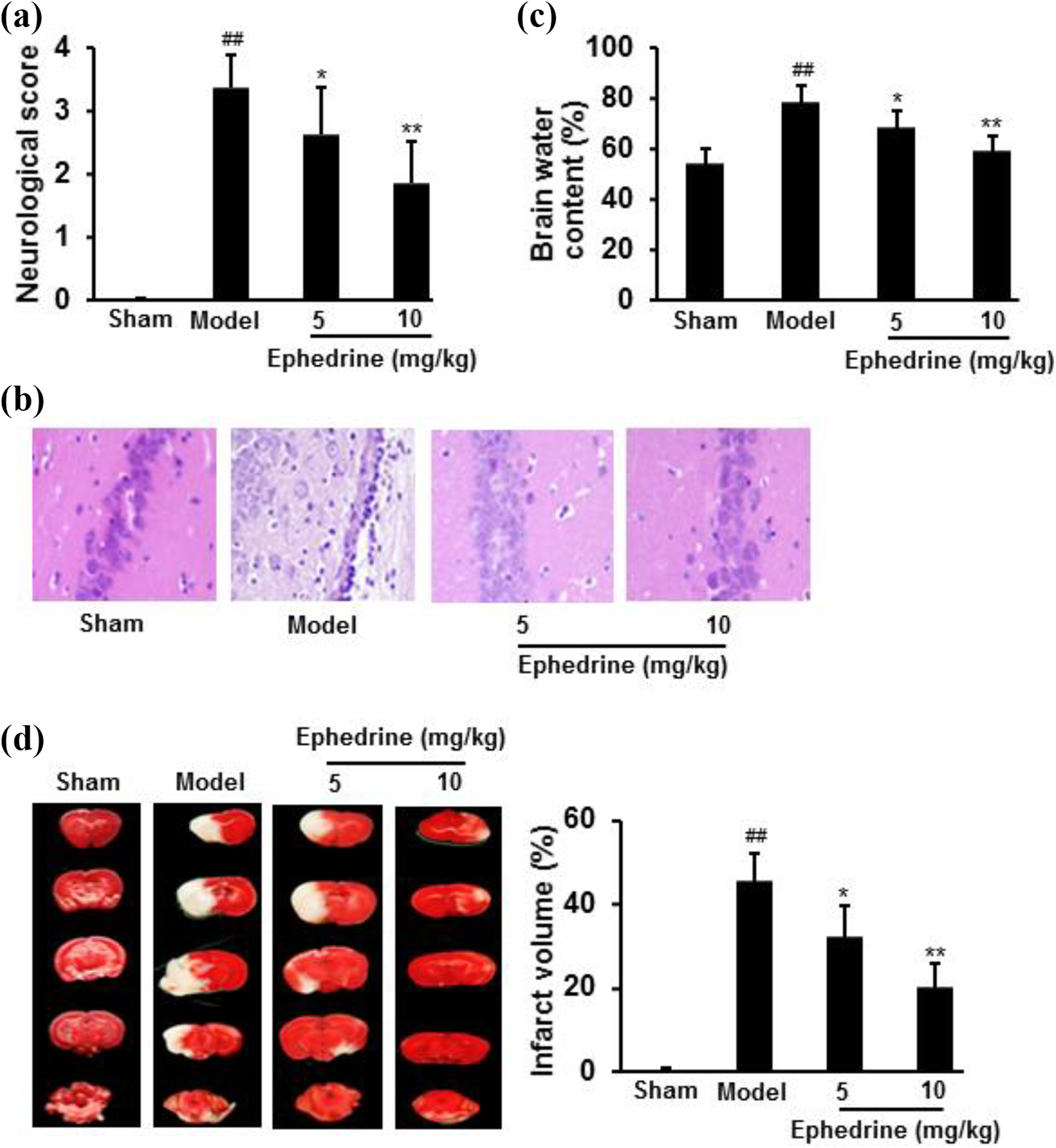
Ephedrine treatment significantly ameliorated brain damage caused by I/R, shown by neurological deficit score (a); brain sections with H&E staining (b); brain water content (c); TTC staining and cerebral infarct size (d). Data were expressed as mean ± SD (n = 20 for a; n = 5 for b–d). ## p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. Model group.
Ephedrine treatment decreased the autophagy in brain tissues
Autophagy in brain tissues was explored by examining the mRNA expression of Beclin-1 and LC3B protein level. Results showed that treatment with ephedrine significantly decreased Beclin-1 mRNA expression and LC3B protein level, compared with I/R group (p < 0.05) (Figure 2).

Ephedrine treatment decreased autophagy in brain tissues induced by I/R. Brain tissues were collected to measure the mRNA and protein expression: Beclin-1 mRNA expression (a) and LC3B protein expression (b). Data were expressed as mean ± SD (n = 5). ## p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. model group.
Ephedrine treatment decreased apoptosis in brain tissues
Compared with sham group, apoptosis distinctly increased in brain tissues of MCAO rats. However, treatment with ephedrine significantly decreased DNA fragmentation, cytochrome-c release and caspase activities (p < 0.05). Moreover, pro-apoptotic protein Bax expression decreased, but anti-apoptotic protein Bcl-2 increased after ephedrine treatment (Figure 3).

Ephedrine treatment decreased apoptosis in brain tissues induced by I/R. Apoptosis was measured by: DNA damage (a); cytochrome-c release (b); caspase activities (c); and apoptosis-related proteins (d). Data were expressed as mean ± SD (n = 5). ## p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. model group.
Ephedrine treatment decreased inflammatory responses in brain tissues
Compared with sham group, levels of IL-1β, IL-6, TNFα and NO significantly increased in brain tissues of the I/R group (p < 0.01). However, levels of these inflammatory factors were significantly lower in the brain tissues after ephedrine treatment (p < 0.05) (Figure 4).

Ephedrine treatment inhibited inflammatory responses in brain tissues. Inflammatory responses were measured by: IL-1β (a); IL-6 (b); TNFα (c) and NO (d). Data were expressed as mean ± SD (n = 5). ## p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. model group.
Ephedrine treatment decreased inflammatory responses through NF-κB Pathway
To further explore the action mechanisms of ephedrine, the relative gene and protein expression levels of inflammatory factors were examined. Results showed that ephedrine treatment significantly decreased the gene expression of IL-1β, IL-6, TNFα and their protein expression, compared with I/R group (p < 0.05). Furthermore, ephedrine treatment decreased the protein expression of p-NF-κB, while not changing NF-κB (Figure 5).

Ephedrine treatment decreased inflammatory responses through NF-κB pathway. Ephedrine treatment decreased the gene expression of IL-1β, IL-6, TNFα (a) and their protein expression (b). Furthermore, ephedrine treatment decreased the protein expression of p-NF-κB, while not changing NF-κB (b). Data were expressed as mean ± SD (n = 5). ## p < 0.01 vs. Sham group; *p < 0.05, **p < 0.01 vs. model group.
Discussion
Ischemic stroke is accompanied by inflammation and oxidative stress. Both in vitro and in vivo ischemic stroke models induced the production of inflammatory mediators, including IL-1β, IL-6, TNFα and NO. Thus, it may be irrefutable to suggest that the inhibition of inflammatory responses is a key strategy to protect the brain from I/R-induced injury. 23 Here we demonstrated that ephedrine treatment attenuated inflammatory responses through inhibiting NF-κB activation, and significantly ameliorated brain damage, shown by decreased neurological deficit score, reduced cerebral infarct size as well as water content in a rat model of transient focal cerebral I/R.
Autophagy plays an essential role in the homeostasis, which can be activated upon physiological stresses such as cell protection response to intracellular stresses and toxic metabolites. 24 Due to the transport and properties of active proteins after autophagy mitosis, neuronal survival is highly dependent on autophagy under physiological conditions. In the lack of peculiar control, autophagy may stimulate apoptosis and degrade cells by self-digestion and eventually leading to death. Recent studies have shown that acute and severe ischemia may cause excessive autophagy, thereby promoting cell damage and death. 25 In this study, Beclin-1 mRNA and LC3B protein expression was examined. Results showed I/R injury caused a significant increase in the expression of Beclin-1 and LC3B. However, the increase can be largely reversed by ephedrine treatment.
Apoptosis has been implicated in cerebral I/R injury. As a principal member of the Bcl-2 family and an important anti-apoptotic protein, Bcl-2 plays a critical role in inhibiting the action of pro-apoptotic proteins and promoting cellular survival. Bcl-2 overexpression reduced ischemic infarct, and enhanced ischemia-induced striatal neurogenesis. 26 Studies indicated the intricate crosstalk between apoptosis and autophagy. 27 Bcl-2 inhibited Beclin-1-dependent autophagy, thereby functioning both as pro-survival and anti-autophagy factor. 28 Our results demonstrated that the neuroprotective effects of ephedrine might associate with an attenuation of ischemia-induced apoptosis through upregulation of the Bcl-2 level.
Inflammatory responses have been considered to be the critical element in the pathological progression of cerebral I/R. 29 The pro-inflammatory signals from immune mediators rapidly activate resident cells and influence infiltration of a wide range of inflammatory cells (monocytes/macrophages, neutrophils, different subtypes of T cells, and other inflammatory cells) into the ischemic region, thereby exacerbating brain damage. 30 TNFα, a potent pro-inflammatory cytokine, is upregulated in the brain after MCAO. TNFα mediates apoptotic signals and inflammatory responses via NF-κB. 31 IL-1β is a key contributor to ischemic brain injury. IL-1β knockout mice have markedly reduced brain damage induced by I/R and the increased brain damage occurred when IL-1β was administered to rats. 32,33 NF-κB has been considered as a prototypical pro-inflammatory signaling pathway. During inflammation, intracellular responsive cascades such as NF-κB become activated to increase expression of pro-inflammatory products. 34,35 In this study, ephedrine treatment significantly reduced levels of inflammatory factors and controlled inflammatory responses, suggesting that the inhibitory effects of ephedrine on the pro-inflammatory factors may be one of the crucial mechanisms in its effects against cerebral I/R injury.
In conclusions, our results suggested that cerebral I/R injury coincided with pathology associated changes in autophagy, apoptosis and inflammatory responses. Ephedrine treatment protected rats against cerebral I/R injury. The mechanism by which ephedrine exerted its neuroprotective activities, at least partly, was associated with inhibiting the processes of inflammation. These data suggested that ephedrine could be further developed as a clinical neuroprotective candidate for treatment of ischemic stroke.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.