
Abstract
Amlodipine-induced toxicity has detrimental effects on cardiac cells. The aim of this study was to examine the effect of lipid emulsion on decreased H9c2 rat cardiomyoblast viability induced by amlodipine toxicity. The effects of amlodipine, lipid emulsion, LY 294002, and glibenclamide, either alone or in combination, on cell viability and count, apoptosis, and expression of cleaved caspase-3 and -8, and Bax were examined. LY 294002 and glibenclamide partially reversed lipid emulsion-mediated attenuation of decreased cell viability and count induced by amlodipine. Amlodipine increased caspase-3 and -8 expression, but it did not alter Bax expression. LY 294002 and glibenclamide reversed lipid emulsion-mediated inhibition of cleaved caspase-3 and -8 expression induced by amlodipine. Lipid emulsion inhibited early and late apoptosis induced by amlodipine. LY 294002 and glibenclamide inhibited lipid emulsion-mediated inhibition of late apoptosis induced by amlodipine, but they did not significantly alter lipid emulsion-mediated inhibition of early apoptosis induced by amlodipine. Lipid emulsion decreased amlodipine-induced TUNEL-positive cells. These results suggest that lipid emulsion inhibits late apoptosis induced by amlodipine at toxic dose via the activation of phosphoinositide-3 kinase and ATP-sensitive potassium channels in the extrinsic apoptotic pathway.
Introduction
Lipid emulsion, used for parenteral nutrition and dissolution of intravenous anesthetics propofol and etomidate, can treat cardiovascular collapse induced by local anesthetics, such as bupivacaine, ropivacaine, mepivacaine, and levobupivacaine, at toxic doses. 1 In addition, several case reports in humans suggest that lipid emulsion ameliorates intractable cardiovascular collapse induced by amlodipine, a long-acting dihydropyridine calcium channel blocker, at toxic doses. 2 –5 The results of an in vivo study showed that lipid emulsion reduced hemodynamic instability and enhanced survival in animals with amlodipine toxicity. 6 In vitro studies have suggested that lipid emulsion inhibits severe vasodilation induced by calcium channel blockers, including amlodipine, bepridil, and verapamil, at toxic doses. 7,8 However, some case reports have suggested that lipid emulsion may have no effect on cardiovascular collapse induced by amlodipine toxicity. 9,10 Overall, the conclusions on lipid emulsion treatment for amlodipine toxicity are conflicting. 2 –5,9,10 Amlodipine toxicity results in a negative inotropic effect and bradycardia in isolated heart preparation, and apoptosis in uveal malignant melanoma and human fibroblasts. 11 –14 A previous study showed that lipid emulsion attenuated apoptosis in cardiomyoblasts induced by the phenylalkylamine calcium channel blocker verapamil at toxic doses. 15 It has been reported that phosphoinositide-3 kinase and ATP-sensitive potassium channels are involved in cardiac protection. 16,17 However, the effect of lipid emulsion on the decreased viability of cardiomyoblasts, induced by amlodipine toxicity, remains unknown. Thus, the aim of this study was to examine the effect of lipid emulsion (Intralipid) on amlodipine induced-apoptosis and elucidate the associated cellular mechanism.
Materials and methods
Cell culture
H9c2 rat embryonic ventricular myocardial cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Life Technologies, Grand Island, NY, USA), containing 10% fetal bovine serum (ATCC), 100 IU/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco), as described previously. 18 The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and were subcultured after reaching around 80% confluence. The cells were starved in serum-free medium for 15 h before drug treatment.
Materials
Amlodipine, LY 294002, glibenclamide, and anti-β actin antibody were obtained from Sigma Aldrich (St. Louis, MO, USA). Intralipid (20%) was obtained from Fresenius Kabi Korea (Seoul, Korea). Anti-Bax antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-cleaved caspase-3 and anti-cleaved caspase-8 antibodies were obtained from Cell Signal Technology (Beverly, MA, USA). Amlodipine and glibenclamide were dissolved in dimethyl sulfoxide (final dimethyl sulfoxide concentration = 0.1%).
Cell morphological analysis
H9c2 cells (1 × 105 cells/mL) were seeded in six-well plates, and subsequently treated with amlodipine, lipid emulsion, LY 294002, and glibenclamide, either alone or in combination. Based on previous studies, the phosphoinositide-3 kinase inhibitor LY 294002 (10−5 M) and ATP-sensitive potassium channel inhibitor glibenclamide (5 × 10−6 M) were used to investigate the role of phosphoinositide-3 kinase and ATP-sensitive potassium channels in the lipid emulsion-mediated protective effect against amlodipine-induced decrease in cell viability. 16,17 The treated cells were washed twice with phosphate-buffered saline (PBS), and their morphology was observed under a phase-contrast microscope (Olympus Optical Co., Ltd., Fluoview™ 500, Tokyo, Japan).
Cell viability assay
Cell Counting Kit 8 (CCK-8) (Dojindo Molecular Technologies, Kumamoto, Japan), a sensitive colorimetric assay kit for determining the number of viable cells, was used to determine cell viability, according to the manufacturer’s instructions. CCK-8 uses WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), which is bio-reduced by cellular dehydrogenases to an orange formazan product that is soluble in culture medium. The amount of formazan produced is directly proportional to the number of living cells. Briefly, the cells were seeded into 24-well plates (2 × 104 cells/well). After treatment with toxic dose (10− 6 to 10− 4 M) of amlodipine, lipid emulsion, LY 294002, and glibenclamide, alone or in combination, DMEM (500 μL), containing 10% CCK-8 solution, was added to each well and the cells were incubated for an additional 3 h at 37°C in a 5% CO2 atmosphere. 19 The optical density of sample in each well was measured at a wavelength of 450 nm using the VersaMax® microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cell-count assay
A cell-count assay was conducted to confirm the result obtained from the cell viability assay showing the protective effect of lipid emulsion on the decreased cell viability induced by toxic dose of amlodipine in H9c2 cells. The cell-count assay was performed as described previously. 18 Briefly, H9c2 cells were seeded into 24-well plates (2 × 104 cells/well). After treatment with amlodipine, lipid emulsion, LY 294002, and glibenclamide, alone or in combination, the cells were harvested with 0.05% trypsin/EDTA and mixed with trypan blue. Cell density was measured using the Countess II FL automated cell counter (Life Technologies, Bothell, WA, USA).
Western blotting
Protein expression levels were determined using western blotting as described by Lee et al. 20 The treated cells in each group were washed twice with ice-cold PBS, and then lysed in ice-cold lysis buffer (Sigma, St. Louis, MO, USA), containing protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL, USA) and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The lysates were centrifuged at 20,000 × g for 15 min at 4°C. Next, the supernatants were collected and the protein content was quantified using the Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA, USA). Total protein (30 μg) was denatured by adding 5× sodium dodecyl sulfate (SDS) sample buffer (0.1 M Tris-HCl, 20% glycerol, 4% SDS, and 0.01% bromophenol blue) and heating at 100°C for 10 min. The sample proteins were then separated using either 12% or 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The separated proteins were transferred onto polyvinylidene difluoride membrane (Immobilon-P, Millipore. Co., Bedford, MA, USA) and were subsequently blocked with 5% bovine serum albumin or 5% skim milk at room temperature for 1 h. The membranes were then incubated overnight with primary antibodies (anti-cleaved caspase-3 [1:500], anti-cleaved caspase-8 [1:500], and anti-Bax [1:1,000]) at 4°C. After washing with Tris-buffered saline with 0.1% Tween 20 (TBST, pH 7.6), the membranes were incubated with the corresponding anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies (diluted to 1:2,000 or 1:5,000) for 1 h at room temperature. After washing thrice with TBST, the immune complexes were visualized with enhanced chemiluminescence (SuperSignal® West Pico Chemiluminescent Substrate, Thermo Scientific). The signals were captured using the ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, USA) and the intensity of the bands was quantified using Image Lab™ Software v.3.0 (Bio-Rad Laboratories).
Annexin V-FITC-PI staining
Amlodipine-induced apoptosis of H9c2 cells was measured by flow cytometry, using the FITC Annexin V Apoptosis Detection Kit (Invitrogen-Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s protocol. 21 Briefly, the cells were cultured in six-well plates (1 × 105 cells/well). After treatment with amlodipine, lipid emulsion, LY 294002, and glibenclamide, alone or in combination, adherent and floating cells were harvested and washed twice with pre-cold PBS, and then resuspended in 1× binding buffer. The suspended cells were then stained with Annexin V-FITC and propidium iodide (PI) working solution (100 μg/mL) in dark for 15 min at room temperature. The cells were then resuspended in 400 µL of 1× binding buffer and immediately analyzed by flow cytometry. The cell viability and apoptosis rates were measured using the FC-500 flow cytometer (Beckman Coulter, Pasadena, CA, USA), with 488-nm laser excitation and fluorescence emission at 530 (FL1) and >575 nm (FL3). A total of 20,000 cells were collected and data from three independent experiments were analyzed, using Beckman Coulter CPX software (Beckman Coulter, CXP 2.2; Mervue Business Park, Mervue, Galway, Ireland). Fluorescence distribution was displayed as a dot plot and the percentage of fluorescent cells in each quadrant was determined.
Terminal deoxynucleotidyl transferase dUTP nick end-labeling assay
Cell apoptosis was detected and quantified using the terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay, with a TUNEL Kit (Roche Applied Science, Indianapolis, IN, USA), according to the manufacturer’s protocol. 18 The nuclei in apoptotic and non-apoptotic cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Chemical Company, St. Louis, MO, USA). Fluorescence staining was observed under a fluorescence microscope (Olympus Optical Co., Ltd., Fluoview 500, Tokyo, Japan). The percentage of apoptotic cells was calculated as the ratio of the number of TUNEL-positive cells to the total number of DAPI-stained cells, counted in three different random fields.
Determination of amlodipine concentration in Krebs solution
Amlodipine (10−5 M) was emulsified with lipid emulsion (Intralipid: 0%, 0.125%, and 0.25%) in Krebs solution, using a rotator for 30 min, as described previously. 7,8 To determine the concentration of amlodipine released from the samples, the emulsified samples were centrifuged at 75,000 × g for 40 min. The concentration of amlodipine in the aqueous phase (equivalent to centrifuged aqueous extract) was measured by ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF MS; Waters, Milford, MA, USA), using an Acquity UPLC BEH C18 column (100 mm × 2.1 mm, 1.7 μm; Waters). The aqueous layer was injected into the column equilibrated with water/acetonitrile (99:1) (containing 0.1% formic acid) and eluted with a linear gradient (1%–100%) of acetonitrile (containing 0.1% formic acid) at a flow rate of 0.35 mL/min for 5 min. The eluted amlodipine was analyzed by Q-TOF MS with multiple reaction monitoring and positive electrospray ionization mode. The capillary and sampling cone voltages were set at 3 kV and 30 V, respectively. The desolvation and source temperatures were set at 100°C and 400°C, respectively, whereas the desolvation flow rate was 800 L/h. The eluted amlodipine was monitored by the precursor and product ions of m/z 409.14 and 294.08, respectively. LockSpray with leucine-enkephalin ([M+H] = 556.2771 Da) was used to ensure the reproducibility and accuracy of all analyses. All mass data were collected and analyzed using UIFI 1.8.2 (Waters).
Statistical analysis
All data are expressed as mean ± standard deviation (SD). The effects of amlodipine, lipid emulsion, LY 294002, and glibenclamide, alone or in combination, on cell viability and count, apoptotic factors (caspase-3, caspase-8, and Bax expression, which are involved in early and late apoptosis), TUNEL-positive cells, and amlodipine concentration were analyzed using the one-way analysis of variance followed by Bonferroni’s multiple comparison test using Prism 5.0 (GraphPad Software, San Diego, CA, USA). The results with a p value of less than 0.05 were considered statistically significant.
Results
Effect of lipid emulsion on the decreased viability of H9c2 rat cardiomyoblasts induced by amlodipine at toxic doses
Amlodipine (3 × 10−6 to 10− 4 M) reduced the viability of H9c2 rat cardiomyoblasts (Figure 1A; p < 0.001 vs. control). Lipid emulsion (0.125% and 0.25%) inhibited the decrease in cell viability induced by amlodipine (10−5 M) (Figure 1B; p < 0.001 vs. amlodipine alone). Lipid emulsion (0.25%) alone decreased cell viability (Figure 1B; p < 0.05 vs. control). In addition, lipid emulsion (0.125% and 0.25%) attenuated the decreased cell count induced by amlodipine (10−5 M) (Figure 1C; p < 0.001 vs. amlodipine alone). Lipid emulsion (0.125% and 0.25%) alone resulted in decreased cell count (Figure 1C; p < 0.05 vs. control). The morphology of H9c2 cells treated with amlodipine, Iipid emulsion, LY 294002, and glibenclamide, alone or in combination, was observed using a phase-contrast microscope (Figure 2A). Control H9c2 cells showed almost no abnormal finding and appeared fusiform and elongated (Figure 2A). Amlodipine (10− 5 M) treatment induced distinct morphological changes, including detachment, shrinkage, and irregular shape (Figure 2A). However, pretreatment with lipid emulsion (0.125%) protected cells from morphological changes induced by amlodipine (10− 5 M) (Figure 2A). LY 294002 (10− 5 M) and glibenclamide (5 × 10− 6 M) inhibited the lipid emulsion (0.125%)-mediated protective effect of morphological changes, induced by amlodipine (10− 5 M) (Figure 2A). Pretreatment with the phosphoinositide-3 kinase inhibitor LY 294002 (10−5 M) or ATP-sensitive potassium channel inhibitor glibenclamide (5 × 10− 6 M) inhibited lipid emulsion-mediated attenuation of decreased cell viability and cell count induced by amlodipine (10− 5 M) (Figures 2B and 2C; p < 0.001 vs. lipid emulsion plus amlodipine).
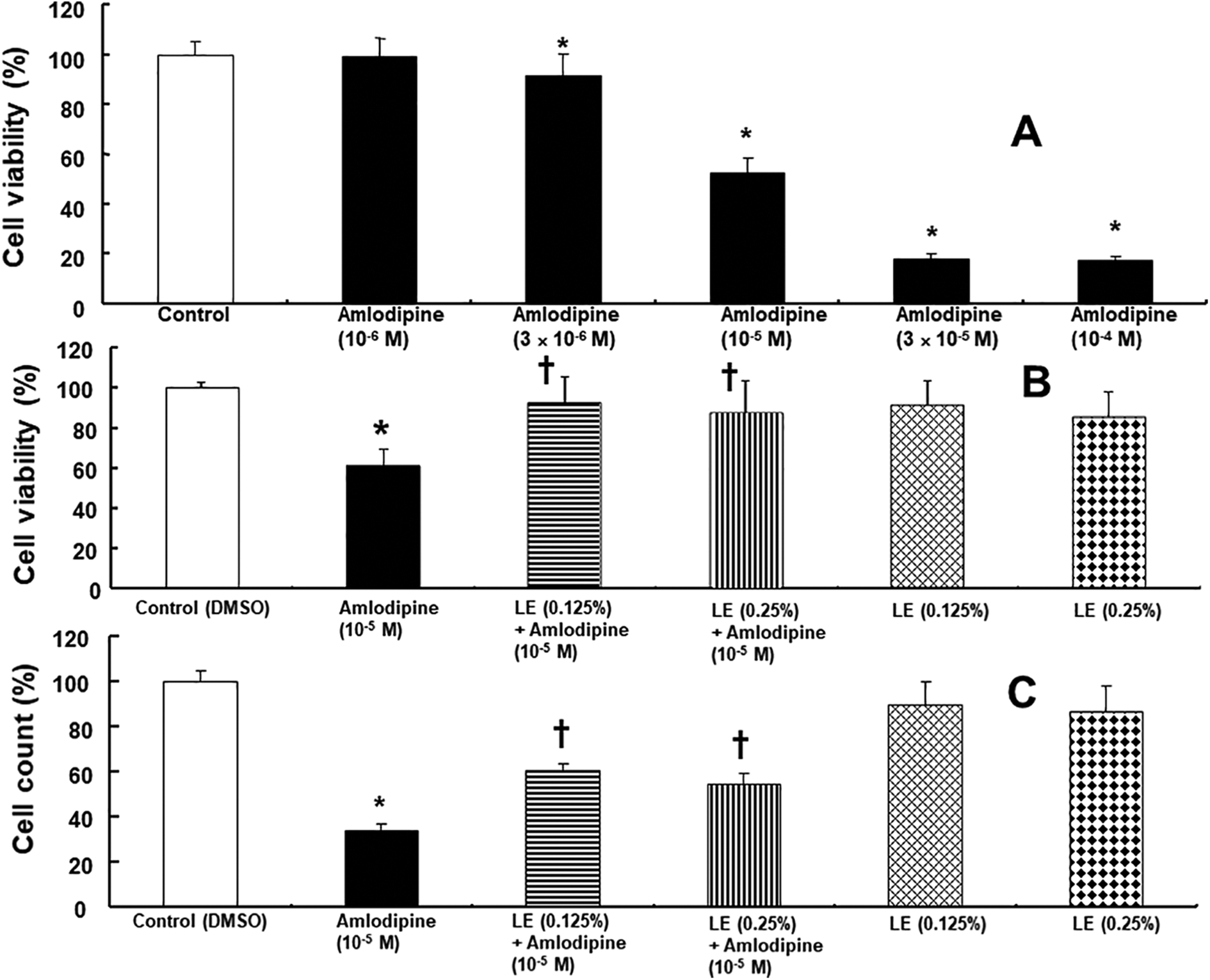
A: Effect of amlodipine alone (n = 20) on the viability of H9c2 rat cardiomyoblasts. H9c2 cells were treated with various concentrations of amlodipine for 24 h. B and C: The effect of lipid emulsion (LE) on decreased viability (n = 12) and count (n = 12) of H9c2 cells induced by amlodipine (10−5 M). H9c2 cells were treated with 10−5 M amlodipine alone for 24 h or pretreated with LE (0.125 and 0.25%) for 1 h, followed by treatment with amlodipine (10−5 M) for 24 h or LE (0.125 and 0.25%) alone for 25 h. Data are shown as mean ± SD of three independent experiments. n indicates the number of samples. DMSO: dimethyl sulfoxide (0.1%). A: *p < 0.001 versus control. B: *p < 0.001 versus control. †p < 0.001 versus amlodipine alone. C: *p < 0.001 versus control. †p < 0.001 versus amlodipine alone.

A: Representative phase-contrast microscopic images of H9c2 cells after treatment with amlodipine, lipid emulsion (LE), LY 294002, and glibenclamide, alone or in combination. H9c2 cells were treated with 10−5 M amlodipine alone for 24 h or pretreated with LE (0.125%) for 1 h, followed by treatment with amlodipine (10−5 M) for 24 h, pretreatment with the inhibitor LY 294002 (10−5 M) or glibenclamide (5 × 10−6 M) for 1 h, followed by treatment with LE for 1 h; the cells were subsequently treated with amlodipine (10−5 M) for 24 h, LE alone for 25 h, or inhibitor (LY 294002 or glibenclamide) alone for 26 h. Scale bar: 100 μm. B and C: Effect of amlodipine, LE, and inhibitors (LY 294002 or glibenclamide), alone or in combination, on the viability (n = 12) and count (n = 12) of H9c2 cells. Data are shown as mean ± SD of three independent experiments. n indicates the number of samples. DMSO: dimethyl sulfoxide (0.1%). *p < 0.001 versus control. †p < 0.001 versus amlodipine alone. #p < 0.001 versus LE + amlodipine.
Lipid emulsion attenuated late apoptosis induced by amlodipine at toxic doses in H9c2 rat cardiomyoblasts
Apoptosis assay was performed to evaluate whether lipid emulsion-mediated attenuation of decreased cell viability induced by toxic dose of amlodipine is associated with apoptosis. Apoptosis is characterized by cell shrinkage, membrane blebbing, chromosomal condensation, nuclear fragmentation, DNA laddering, and finally phagosome engulfment. Immediately following apoptosis initiation, cells displace membrane phosphatidyl serine (PS) from the inside of the plasma membrane to the surface, and then DNA is fragmented. Therefore, we observed cell surface PS and DNA damage by staining with an affinity protein Annexin V and PI, respectively. Moreover, TUNEL assay was conducted to assess DNA fragmentation.
Amlodipine (10−5 M) upregulated the expression of cleaved caspase-3 (Figure 3A; p < 0.001 vs. control), whereas lipid emulsion (0.125%) inhibited amlodipine (10−5 M)-induced increase in caspase-3 expression (Figure 3A; p < 0.001 vs. amlodipine alone). LY 294002 (10−5 M) and glibenclamide (5 × 10−6 M) exerted an inhibitory effect on the lipid emulsion (0.125%)-induced decrease in cleaved caspase-3 expression, induced by amlodipine (10−5 M) (Figure 3A: p < 0.01 vs. lipid emulsion plus amlodipine). Amlodipine (10−5 M) upregulated the expression of the extrinsic pro-apoptotic protein cleaved caspase-8 (Figure 3B; p < 0.001 vs. control), however, lipid emulsion (0.125%) decreased this effect (Figure 3B; p < 0.001 vs. amlodipine alone). LY 294002 (10−5 M) and glibenclamide (5 × 10−6 M) inhibited lipid emulsion (0.125%)-mediated attenuation of cleaved caspase-8 expression, induced by amlodipine (Figure 3B; p < 0.05 vs. lipid emulsion plus amlodipine). However, amlodipine (10−5 M) did not significantly alter the expression of the intrinsic proapoptotic protein Bax (Figure 4). Treatment with lipid emulsion (0.125%), either alone or in combination with either LY 294002 (10−5 M) or glibenclamide (5 × 10−6 M), did not alter amlodipine (10−5 M)-induced Bax expression significantly (Figure 4). In addition, lipid emulsion (0.125%), LY 294002 (10−5 M), and glibenclamide (5 × 10−6 M) alone did not significantly alter the expression of cleaved caspase-3, caspase-8, and Bax (Figures 3 and 4). Amlodipine (10−5 M) increased the percentage of early and late apoptosis (Figure 5; p < 0.001 vs. control). On the contrary, lipid emulsion (0.125%) inhibited the percentage of early and late apoptosis, induced by amlodipine (10−5 M) (Figure 5; p < 0.001 vs. amlodipine alone). LY 294002 (10−5 M) and glibenclamide (5 × 10−6 M) attenuated lipid emulsion-mediated inhibition of late apoptosis, induced by amlodipine (10−5 M) (Figure 5B; p < 0.001 vs. lipid emulsion plus amlodipine). However, LY 294002 and glibenclamide did not significantly alter lipid emulsion (0.125%)-mediated inhibition of early apoptosis, induced by amlodipine (10−5 M) (Figure 5B). Lipid emulsion (0.125%), LY 294002 (10−5 M), and glibenclamide (5 × 10−6 M) alone did not significantly alter early and late apoptosis (Figure 5). Furthermore, amlodipine (10−5 M) increased the percentage of TUNEL-positive cells (Figure 6; p < 0.001 vs. control), whereas lipid emulsion (0.125%) inhibited this effect (Figure 6; p < 0.001 vs. amlodipine alone).

A and B: Effect of amlodipine, lipid emulsion (LE), and inhibitors (LY 294002 and glibenclamide), alone or in combination, on the expression of cleaved capase-3 and cleaved caspase-8. H9c2 cells were treated with 10−5 M amlodipine alone for 12 h or pretreated with LE (0.125%) for 1 h, followed by treatment with amlodipine (10−5 M) for 12 h, pretreated with inhibitor (10−5 M LY 294002 or 5 × 10−6 M glibenclamide) for 1 h, followed by treatment with LE for 1 h; and subsequently treated with amlodipine (10−5 M) for 12 h, LE (0.125%) alone for 13 h, or inhibitor alone for 14 h. Data (n = 4) are shown as mean ± SD. n indicates the number of independent experiments. DMSO: dimethyl sulfoxide (0.1%). *p < 0.001 versus control. †p < 0.001 versus amlodipine alone. §p < 0.05 and #p < 0.01 versus LE + amlodipine.

Effect of amlodipine, lipid emulsion (LE), and inhibitor (LY 294002 or glibenclamide), alone or in combination, on Bax expression. H9c2 cells were treated with 10−5 M amlodipine alone for 12 h or pretreated with LE (0.125%) for 1 h followed by amlodipine (10−5 M) for 12 h, pretreated with inhibitor (10−5 M LY 294002 or 5 × 10−6 M glibenclamide) for 1 h, followed by treatment with LE for 1 h; the cells were subsequently treated with amlodipine (10−5 M) for 12 h, LE alone for 13 h, or inhibitor alone for 14 h. Data (n = 4) are shown as mean ± SD. n indicates the number of independent experiments. DMSO: dimethyl sulfoxide (0.1%).

A: Effects of lipid emulsion (LE) on the amlodipine-induced apoptosis of H9c2 cells. H9c2 cells were treated with 10−5 M amlodipine alone for 12 h or pretreated with LE (0.125%) for 1 h, followed by amlodipine (10−5 M) for 12 h, pretreated with inhibitor LY 294002 (10−5 M) or glibenclamide (5 × 10−6 M) for 1 h, followed by LE for 1 h; subsequently, the cells were treated with amlodipine (10−5 M) for 12 h, LE alone for 13 h, or inhibitor alone for 14 h. Annexin V/ propidium iodide staining followed by flow cytometric analysis. B: Plot (n = 3) showing both early- and late-stage apoptosis after treatment. Data are shown as mean ± SD. n indicates the number of independent experiments. DMSO: dimethyl sulfoxide (0.1%). *p < 0.001 versus control. †p < 0.001 versus amlodipine alone. #p < 0.01 and §p < 0.05 versus LE + amlodipine.
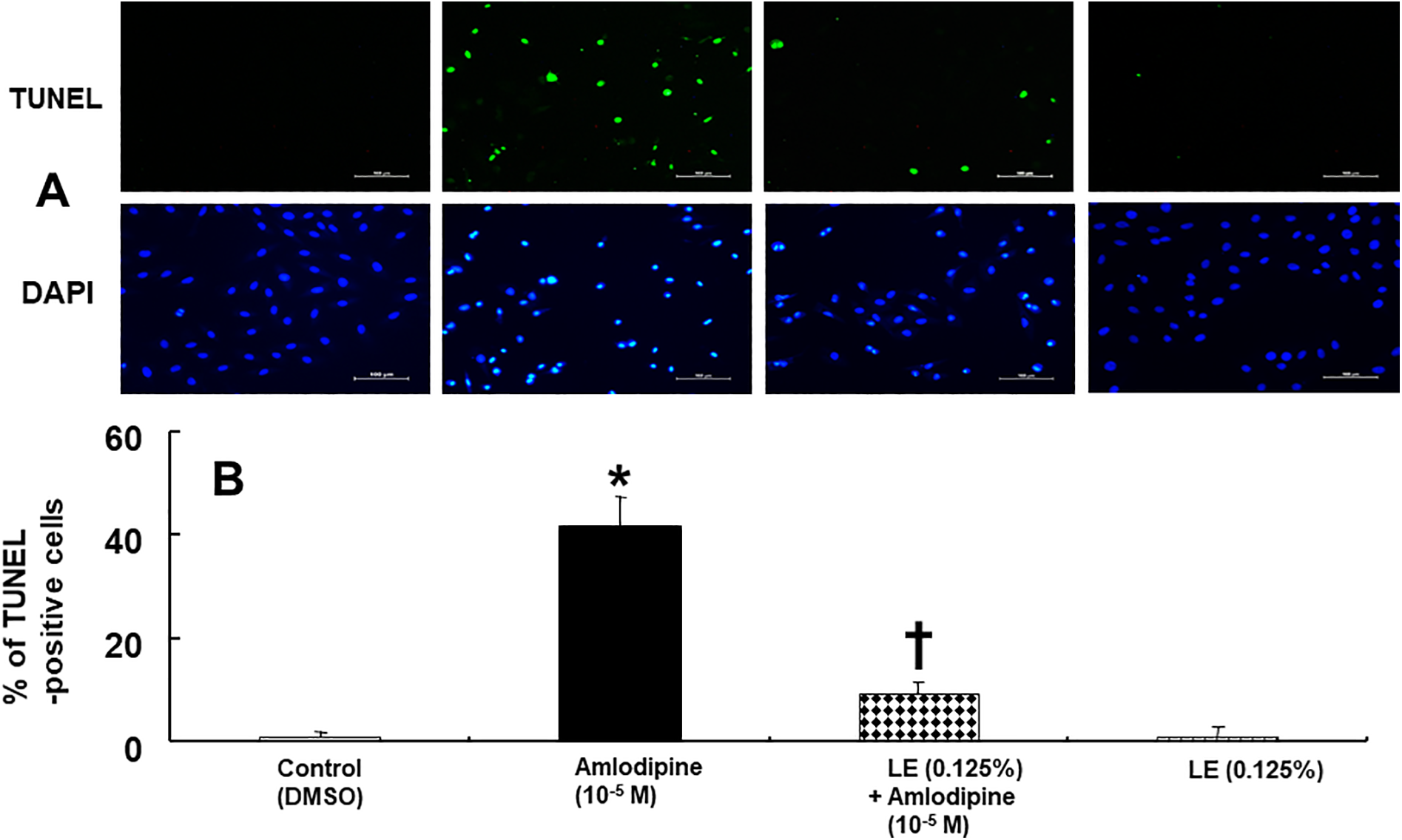
TUNEL staining of H9c2 cells treated with amlodipine or lipid emulsion (LE) alone or with LE combined with amlodipine. A: H9c2 cells were treated with 10−5 M amlodipine alone for 24 h or LE (0.125%) alone for 25 h or 10−5 M amlodipine for 24 h, after pretreatment with LE (0.125%) for 1 h. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and are shown in blue. B: Percent of TUNEL-positive H9c2 cells treated with control, amlodipine or LE alone and combined treatment with LE and amlodipine. Data are shown as mean ± SD (n = 3). n indicates the number of independent experiments. *p < 0.001 versus control. †p < 0.001 versus amlodipine alone. DMSO: dimethyl sulfoxide (0.1%). Scale bar: 100 μm
Effect of lipid emulsion on amlodipine concentration in the Krebs solution
Lipid emulsion (0.125% and 0.25%) slightly decreased amlodipine (10−5 M) concentration (Figure 7; P < 0.01 vs. amlodipine alone) in the Krebs solution.

Effect of lipid emulsion (LE) on the amlodipine concentration (10−5 M) in Krebs solution. Amlodipine concentration was measured by ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Data are shown as mean ± SD and expressed as the percentage of the original amlodipine concentration (10−5 M). Experiments were repeated five times. *p < 0.01 versus amlodipine alone.
Discussion
To the best of our knowledge, this is the first study to suggest that lipid emulsion attenuates late apoptosis induced by amlodipine toxicity via the activation of phosphoinositide-3 kinase and ATP-sensitive potassium channels in rat cardiomyoblasts. The major findings of this study were as follows: 1) lipid emulsion attenuated amlodipine-induced decrease in cell viability; 2) LY 294002 and glibenclamide partially reversed lipid emulsion-mediated inhibition of increased caspase-3 and -8 expression, induced by amlodipine; 3) LY 294002 and glibenclamide partially reversed lipid emulsion-mediated attenuation of amlodipine-induced late apoptosis; and 4) lipid emulsion reduced the percentage of TUNEL-positive cells, which were increased by amlodipine.
Amlodipine at toxic doses (3 × 10−6 to 10− 4 M) decreased cell viability in a dose-dependent manner (Figure 1A). Previous studies have demonstrated that lipid emulsions, such as SMOFlipid® (Fresenius Kabi Austria GmbH, Graz, Austria) and Intralipid, inhibit the reduced viability of H9c2 cardiomyoblasts induced by bupivacaine, verapamil, and doxorubicin at toxic doses. 15,18,21 In line with the findings of previous studies, lipid emulsion (0.125% and 0.25%) attenuated the decreased cell viability and count, caused by amlodipine at a toxic dose (10−5 M). 15,18,19,21 A previous study has also showed that treatment with lipid emulsion attenuates cardiac reperfusion injury via the pathway involving phosphoinositide-3 kinase, Akt, and glycogen synthase kinase-3β phosphorylation. 22 In addition, the activation of ATP-sensitive potassium channels has been implicated in cardiac protection from high glucose-induced injury in H9c2 cardiomyoblasts. 16,23 An anticholinergic drug, penehyclidine, attenuated anoxia/reoxygenation injury in H9c2 cells via the activation of ATP-sensitive potassium channel and pathways involving phosphoinositide-3 kinase/Akt and glycogen synthase kinase-3β. 17 Moreover, lipid emulsion and linoleic acid alone enhanced ATP-sensitive potassium channel activity in HEK-293 cells, transfected with Kir6.2 and SUR2. 24 In our study, the phosphoinositide-3 kinase inhibitor LY 294002 and ATP-sensitive potassium channel inhibitor glibenclamide partially reversed lipid emulsion-mediated attenuation of decreased cell viability and cell count induced by amlodipine at toxic doses (Figure 2B and 2C). Taking into consideration data from previous studies, our results suggest that lipid emulsion-mediated protection from decreased cell viability seems to be partially mediated via the activation of phosphoinositide-3 kinase and ATP-sensitive potassium channels in cardiomyoblasts. 16,17,22 –24 It has been reported that lipid emulsion inhibits the expression of cleaved caspase-8 and/or Bax induced by bupivacaine, verapamil, doxorubicin, and malathion at toxic doses in rat cardiomyoblasts and lung tissue. 15,18,21,25,26 In the present study, lipid emulsion inhibited amlodipine (10−5 M)-induced increase in cleaved caspase-3 and capase-8 expression (Figure 3A and 3B). However, it did not significantly change the expression levels of the intrinsic apoptotic protein Bax induced by amlodipine (Figure 4). Taken together, these results suggest that lipid emulsion inhibits amlodipine-induced apoptosis of cardiomyoblasts via the extrinsic apoptotic pathway. These results were confirmed using the TUNEL assay, which demonstrated that lipid emulsion reduced the percentage of amlodipine-induced TUNEL-positive cells (Figure 6). Consistent with the cell viability assay results, LY 294002 and glibenclamide partially reversed lipid emulsion-mediated inhibition of cleaved caspase-3 and caspase-8 expression induced by amlodipine (Figures 3A and 3B). This indicates that the inhibition of amlodipine-induced extrinsic apoptosis by lipid emulsion may be mediated by a pathway involving phosphoinositide-3 kinase and ATP-sensitive potassium channels. Lipid emulsion inhibited early and late apoptosis induced by amlodipine (Figure 5). However, LY 294002 and glibenclamide reversed lipid emulsion-mediated inhibition of late apoptosis, but had no significant effect on lipid emulsion-mediated inhibition of early apoptosis (Figure 5). Taken together, these results suggest that lipid emulsion-mediated inhibition of late apoptosis, induced by amlodipine, involves the activation of phosphoinositide-3 kinase or ATP-sensitive potassium channel in the extrinsic apoptosis pathway. The interaction between ATP-sensitive potassium channel and connexin43, which contributed to protecting H9c2 cells from hypoxia-induced apoptosis, was reported to be mediated by protein kinase C. 27 Insulin produces an ATP-sensitive potassium channel-induced membrane hyperpolarization in cultured vascular smooth muscle cells via phosphoinositide-3 kinase. 28 Hence, further studies are warranted to examine both upstream and downstream cellular signal pathways, involving phosphoinositide-3 kinase and ATP-sensitive potassium channels associated with lipid emulsion-mediated inhibition of late apoptosis, induced by amlodipine at toxic doses.
Studies have shown that lipid emulsion reduces the concentration of drugs with a high lipid solubility (log [octanol/water] partition coefficient: >2), such as bupivacaine, verapamil, and amlodipine, by sequestrating drug molecules on the lipid phase of the emulsion. 7,8,24 Consistent with these findings of previous studies, lipid emulsion-mediated slight reduction in amlodipine concentration in this study might have partially contributed to the inhibition of amlodipine-induced decrease in cell viability. 7,8,24
Our results suggest that lipid emulsion (0.125%) contributes to cardiac protection via the inhibition of apoptosis of cardiomyoblasts, induced by amlodipine toxicity. However, this study had a few limitations. First, it was an in vitro study; an in vivo model is better suited for the evaluation of amlodipine-induced severe myocardial depression and vasodilation, leading to cardiovascular collapse. 29 In addition, heart hanging in Langendorff perfusion system is a better suited experimental model for examining the effect of lipid emulsion on the direct myocardial depression induced by toxic dose of amlodipine. 30 Second, H9c2 cardiomyoblasts, obtained from the rat heart, have been reported to have characteristics similar to the skeletal muscle. 31 Even with these limitations, this study suggests that lipid emulsion contributes to cardiac protection from apoptosis induced by amlodipine toxicity.
In conclusion, these results suggest that lipid emulsion-mediated inhibition of late apoptosis, increased by amlodipine toxicity via extrinsic apoptosis pathway, seems to be mediated partially by pathways involving phosphoinositide-3 kinase and ATP-sensitive potassium channels.
Footnotes
Authors’ note
Seong-Ho Ok and Seung Hyun Ahn contributed equally to this work and should be regarded as co-first authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07043914 and NRF-2016R1D1A1B03930451).