
Abstract
Diabetic nephropathy (DN) is primary cause of end-stage renal disease. A previous study has shown that miR-32-5p (miR-32) is highly expressed in kidney tissue during chronic allograft dysfunction with interstitial fibrosis and tubular atrophy. However, the role of miR-32-5p (miR-32) in DN is still unclear. In this study, streptozotocin-induced DN rat models and high glucose (HG)-incubated human kidney proximal tubular epithelial (HK-2) cells were established to investigate the role and underlying mechanisms of miR-32 in DN. Results of real-time PCR revealed that miR-32 levels were greatly increased in DN rats and HG-incubated HK-2 cells. Downregulation of miR-32 effectively relieved HG-induced autophagy suppression, fibrosis, epithelial-mesenchymal transition (EMT) and inflammation in HK-2 cells. Besides, miR-32 overexpression significantly down-regulated the expression of mothers against decapentaplegic homolog 7 (SMAD7), whereas knockdown of miR-32 markedly up-regulated the level of SMAD7. Dual-luciferase reporter gene assay confirmed that SMAD7 was a target of miR-32. Reintroduction of SMAD7 expression rescued miR-32-induced HK-2 cells autophagy suppression, EMT and renal fibrosis. Our findings indicate that miR-32 may play roles in the progression of EMT and fibrosis in DN.
Keywords
Introduction
Diabetic nephropathy (DN) is one of the most common microvascular complications of diabetes, which is the primary reason of end-stage renal disease worldwide. 1 Approximately 30% of patients with type Ⅰ diabetes and 40% of patients with type Ⅱ diabetes ultimately develop DN. Notably, many macrovascular complications, such as atherosclerosis, hypertension and stroke, are associated with the presence of DN. The mortality caused by DN rose by 94% from 1990 to 2012. 2,3 The typical features of DN include glomerular hyperfiltration and hyperperfusion, mesangial matrix expansion and hypertrophy, glomerular basement membrane thickening, as well as accumulation of extracellular matrix (ECM) proteins, thereby leading to glomerulosclerosis, tubulointerstitial inflammation, fibrosis, and even renal failure. 4 Despite great advances in DN treatment, the pathogenesis of DN is still elusive. Therefore, widespread innovation is necessary to improve health conditions for patients with DN.
microRNAs (miRNAs, miRs) are a class of small noncoding RNAs of approximate 19–24 nucleotides in length. Mature miRNAs bind to the 3′-untranslated regions (3′-UTRs) within target messenger RNAs (mRNAs), thereby directing mRNA degradation and/or inhibiting translation. 5 miRNAs play substantial roles in the cell cycle, proliferation, apoptosis, differentiation and gene regulation. 6 Growing evidence suggests that miRNAs are involved in diabetes. 7,8 miR-32-5p (miR-32) is a kind of miRNA located on chromosome band Xq26.2. Increased expression of miR-32 was found in type Ⅱ diabetes patients with obesity and streptozotocin (STZ)-induced diabetic rats. 9,10 A previous study revealed that miR-32 level was obviously enhanced in kidney tissue during chronic allograft dysfunction with interstitial fibrosis and tubular atrophy, 11 implying that miR-32 may be associated with renal fibrosis. However, the role and underlying mechanisms of miR-32 are elusive in DN.
SMAD7, also namely mothers against decapentaplegic homolog 7, is a key negative regulator of transforming growth factor β (TGFβ) signaling. Multiple studies indicated that SMAD7 exerted a protective role in renal injury. 12 –14 SMAD7 inhibited renal fibrosis by altering expression of TGFβ/SMAD3-regulated miRNAs. 12 Li et al. found that SMAD7 suppressed TGFβ-induced SMAD2 activation with the prevention of collagen synthesis and myofibroblast transformation. 13 Another study suggested that SMAD7 protected against acute kidney injury through rescuing tubular epithelial cells from the G1 cell cycle arrest. 14 SMAD7 has been found to play a protective role in diabetes and diabetes-related complications. 15,16 Overexpression of SMAD7 attenuated kidney injury in a rat model of diabetes. 15 Besides, SMAD7 alleviated TGFβ-mediated inflammation and fibrotic response in diabetic wound skin. 16 However, it remains unclear whether SMAD7 mediates the role of miR-32 in DN.
In this study, STZ-induced DN rats and high glucose (HG)-incubated human kidney proximal tubular epithelial (HK-2) cells models were established to investigate the role and underlying mechanism of miR-32 in DN.
Methods
Animal model
Six-week-old male Sprague-Dawley (SD) rats (Changsheng, Benxi, China) were used for all experiments. All animal procedures followed the guide for the care and use of laboratory animals and this study was approved by the Experimental Animal Ethics Committee of The Fourth Affiliated Hospital of Harbin Medical University. Rats (n = 24) were randomly divided into two groups. The animals of normal control group (NC, n = 8) received a standard diet and the other animals (n = 16) were fed a high-fat diet (HFD) for 8 weeks. HFD-fed rats received 30 mg/kg STZ (S110910, Aladdin, Shanghai, China) in citrate buffer intraperitoneally, while NC rats were injected with an equal volume of citrate buffer. Blood glucose levels were measured 3 days after injection and rats with random glucose levels ≥16.7 mmol/L were regarded as diabetic. 17 After 4 weeks of hyperglycemia, urine samples after 24 h were collected and rats with urinary microalbumin (UMA) levels ≥30 mg/day were considered to have DN. 17
Cell culture
Human kidney proximal tubular epithelial (HK-2) cells were purchased from Procell (Wuhan, China). Cells were cultured in minimum Eagle’s medium (MEM; PM150455, Procell) supplemented with 10% fetal bovine serum (SH30084.03, Hyclone, Logan, Utah, USA) at 37°C and 5% CO2. After serum deprivation for 8 h, cells from the normal glucose (NG) group were cultivated in MEM with 5 mmol/L glucose (PB180418, Procell), and cells from the high glucose (HG) group were exposed to 30 mmol/L glucose for 48 h.
Cell transfection
The hsa-miR-32 agomir, hsa-miR-32 antagomir, and their corresponding negative controls (NC) were obtained from GenePharma (Shanghai, China). HK-2 cells were transfected using Lipofectamine 2000 reagent (11668-019, Invitrogen, Carlsbad, California, USA) according to the manufacturer’s protocol. A SMAD7-overexpression pcDNA3.1 plasmid (Vigene, Jinan, China) was cotransfected into HK-2 cells with a miR-32 agomir. An empty pcDNA3.1 vector was regarded as negative control. After 48 h of transfection, RNA and protein were monitored by quantitative Real-time Polymerase Chain Reaction (qRT-PCR) and western blot.
ELISA
Human IL-1β ELISA Kit or human IL-6 ELISA Kit (Uscn, Wuhan, China) was used to assess the effect of miR-32 on inflammation. The assay was carried out according to the instructions of manufacturer.
Dual-luciferase reporter gene assay
The 3′-UTR segments of SMAD7 containing wild type (WT) or mutated type (Mut) miRNA-responsive elements were inserted into the pmirGLO vector (Promega, Madison, USA): SMAD7 3′-UTR WT: 5′-GCATT…TATTG
qRT-PCR
Total RNA was isolated from the treated tissues and cells with Total RNA Isolating Kit (DP419, Tiangen, Beijing, China) according to the manufacturer’s instructions. Samples of 1 µg total RNA were reverse transcribed into complementary DNA (cDNA) using an RT Primer (Genscript, Nanjing, China) and RNase inhibitor (DP418, Tiangen, Beijing, China). qRT-PCR was carried out using SYBR Green PCR kits (SY1020, Solarbio, Beijing, China) to examine the relative expression levels of miR-32 and SMAD7. The expression of GAPDH was used as an internal control of mRNA level. The expression of U6 was applied as an internal control of miRNA level. The primers are shown in Table 1.
Primers sequenced for PCR.

Western blot
Cells were lysed for 5 min in ice-cold RIPA buffer (R0010, Solarbio, Beijing, China) containing PMSF (P0100, Solarbio). The supernatant was collected after centrifugation at 10000 g for 5 min at 4°C. Equal amounts of protein were separated on 5% sodium dodecyl sulfate-polyacrylamide gel and then transferred onto polyvinylidene fluoride (PVDF; IPVH00010, Millipore, MA, USA) membrane. After blocking in 5% (m/v) skimmed milk (A600669, Sangon Biotech, Shanghai, China) for 1 h at room temperature, the membranes were incubated with primary antibodies against P62 (1: 1000 dilution, #39749, CST, Boston, USA), light chain 3 II/I (LC3 II/I; 1: 1000 dilution, #4108, CST), E-cadherin (1: 500 dilution, #14472, CST), vimentin (1:500 dilution, #5741, CST), α-smooth muscle actin (α-SMA; 1: 500 dilution, 55135-1-AP, Proteintech, Beijing, China), SMAD7 (1: 500 dilution, bs-0566 R, Bioss, Beijing, China), type IV Collagen (Collagen IV; 1:400 dilution, bs-4595 R, Bioss), fibronectin (FN; 1: 400 dilution, bs-0666 R, Bioss), and GAPDH (1: 10000 dilution, 60004-1-Ig, Proteintech) at 4°C overnight. The HRP-conjugated secondary antibodies (SE131/ SE134, Solarbio) at a dilution of 1: 3000 in blocking buffer were incubated for 1 h at 37°C. Blots were then visualized using the ECL system (PE0010, Solarbio). The relative protein levels of interest were normalized to that of GAPDH protein.
Statistical analysis
Data were analyzed using the Graph-pad Prism 8.0. All experiments were performed three times. The unpaired two-tailed Student t-test was used to assess the difference between two groups. For multiple groups’ difference, one-way ANOVA followed by Dunnett test as a post hoc test was used. All results were means the mean ± standard deviation (SD). Values for p ≤ 0.05 were defined as statistically significant.
Results
MiR-32 is upregulated in the kidneys of DN rats
Blood glucose and urinary microalbumin changes in the investigated rats are given in Table 2. After 4 weeks of STZ-induced diabetes, urinary microalbumin had a significant difference between the DN group and the NC group (***p ≤ 0.001), and data revealed a fivefold increase of urinary microalbumin in DN rats (Table 2). As shown in Figure 1(a), miR-32 level was markedly enhanced in kidney of DN rats with respect to NC rats (***p ≤ 0.001). We also found the SMAD7 expression to be greatly downregulated in kidney of DN rats, as evaluated by qRT-PCR (Figure 1(b), ***p ≤ 0.001).
Blood glucose and urinary microalbumin variables in NC and DN rats.a

NC: normal control; DN: Diabetic nephropathy.
a Values are mean ± SD.
bp < 0.001 versus NC rats.

Expression of miR-32 in diabetic nephropathy (DN) rats. (a) miR-32 and (b) SMAD7 expression levels were detected by quantitative real-time Polymerase Chain Reaction (qRT-PCR) in kidney of normal control (NC) rats and DN rats (n = 6/group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
MiR-32 affects fibrosis, EMT, inflammation and autophagy in HG-incubated HK-2 Cells
Having determined that DN induced miR-32 expression in kidney of rats, we assessed whether exposure to HG milieu affected miR-32 expression in HK-2 cells. Compared with exposure to NG, miR-32 content was significantly enhanced in HG milieu (Figure 2(a), ***p ≤ 0.001). MiR-32 was highly expressed in NG-treated HK-2 cells after using hsa-miR-32 agomir transfection (***p ≤ 0.001). However, miR-32 level was markedly lower when HK-2 cells were transfected with a hsa-miR-32 antagomir (Figure 2(b), ***p ≤ 0.001). The results of western blot showed that miR-32 overexpression significantly upregulated the levels of Col IV and FN in HG-incubated HK-2 cells (Figure 2(c), **p ≤ 0.01, ***p ≤ 0.001), while silencing of miR-32 downregulated their levels (Figure 2(d), ***p ≤ 0.001). Changes of EMT markers were further explored in HK-2 cells after HG treatment. E-cadherin expression levels were greatly decreased in HG and hsa-miR-32 agomir (HG + hsa-miR-32 agomir) treatment group but were increased in HG + hsa-miR-32 antagomir group (Figure 2(e) and (f), **p ≤ 0.01, ***p ≤ 0.001). In contrast to the E-cadherin expression, miR-32 overexpression upregulated the expression level of vimentin and α-SMA, and silencing of miR-32 downregulated the vimentin and α-SMA expression. Besides, miR-32 overexpression obviously promoted the protein expression of IL-6, IL-1β and P62 in HG-incubated HK-2 cell, while knockdown of miR-32 significantly inhibited these protein expression (Figure 2(g) to (i), *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001). Furthermore, the ratio of LC3 II to LC3 I greatly decreased after overexpression of miR-32, but the ratio of LC3 II to LC3 I was restored after miR-32 silencing (Figure 2(i), ***p ≤ 0.001).

MiR-32 expression in human kidney proximal tubular epithelial (HK-2) cells. (a) HK-2 cells were exposed to either normal (5 mM NG) or high (30 mM HG) glucose concentrations for 48 h. MiR-32 levels were detected by qRT-PCR. (b) qRT-PCR was used to assess expression of miR-32 transfected with either miR-32 agomir or antagomir. After miR-32 agomir/antagomir transfection, protein expression levels of (c and d) type IV Collagen (Collagen IV) and fibronectin (FN), (e and f) α-smooth muscle actin (α-SMA), E-cadherin and vimentin, (h and i) LC3 II/I and P62 were determined by western blot in HG HK-2 cells. (g) The protein expression levels of IL-6 and IL-1β. Error bars represent SD (n = 3/group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
SMAD7 is a target of miR-32
Next, we assessed the regulation of SMAD7 by miR-32 in HK-2 cells under NG condition. Overexpression of miR-32 by transfection with miR-32 agomir, in comparison to miR-32 agomir NC group, extremely reduced SMAD7 mRNA levels, whereas silencing of miR-32 transfected by miR-32 antagomir obviously enhanced SMAD7 mRNA levels (Figure 3(a), ***p ≤ 0.001). These results were also confirmed in western blot (Figure 3(b) and (c), ***p ≤ 0.001), which supported our assumption that SMAD7 may be a target of miR-32. To confirm these results, we predicted the miR-32 potential binding site through TargetScan (http://www.targetscan.org). The 3′-UTR of SMAD7 exactly matched the sequence of miR-32 (Figure 3(d)). We fused the WT or the MUT 3′-UTR of SMAD7 to a luciferase reporter gene. Luciferase activity was greatly decreased in miR-32 agomir transfected HK-2 cells with constructs containing the WT 3′-UTR of SMAD7 compared with agomir-transfected NC group. However, luciferase activity was not changed when the 3′-UTR of SMAD7 bore mutations at the miR-32 binding site (**p ≤ 0.01).
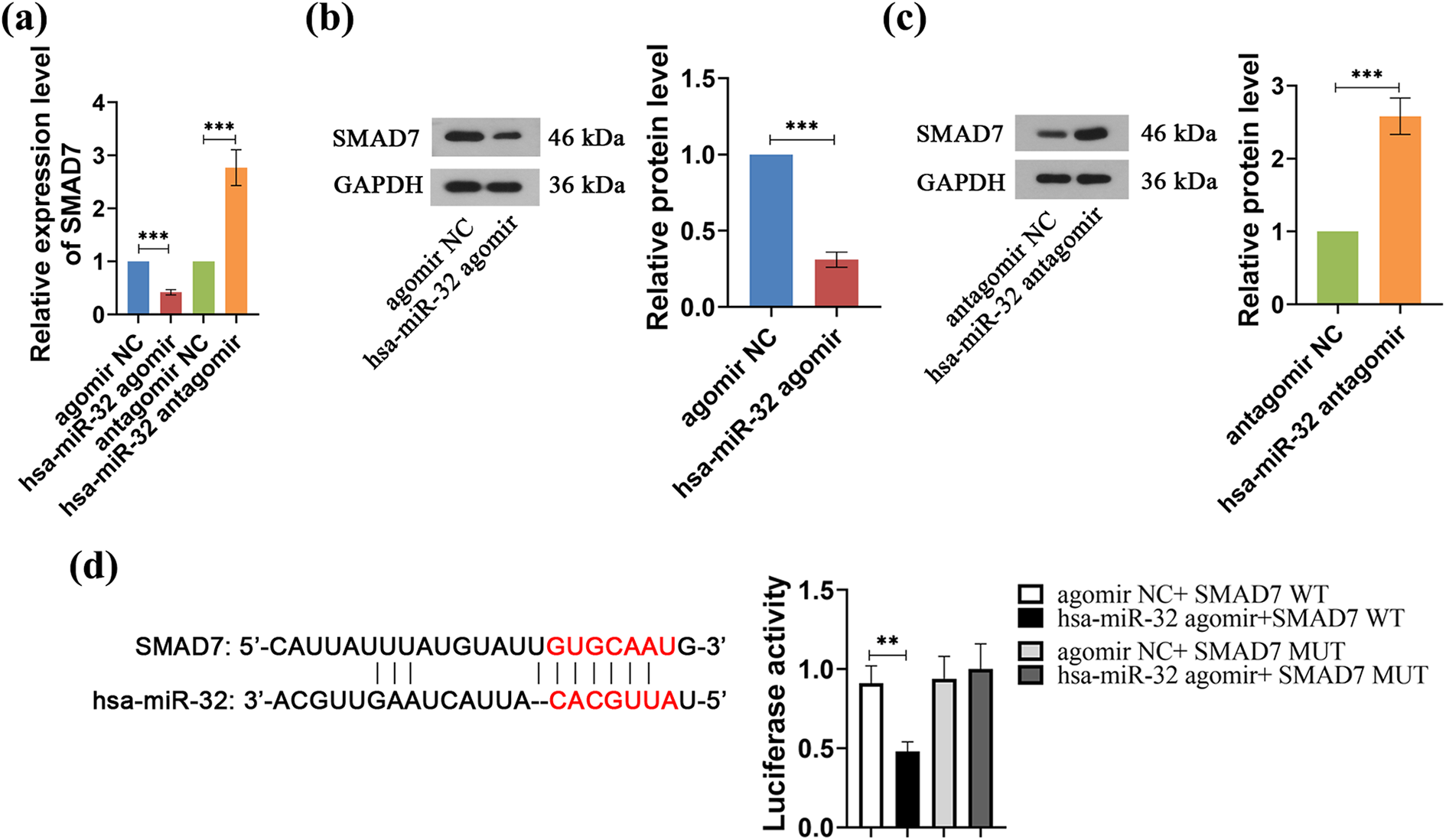
SMAD7 as a target of miR-32 in HK-2 cells exposed to HG. (a) qRT-PCR was used to detect the SMAD7 mRNA level by transfection with miR-32 agomir or antagomir. (b and c) Protein levels of SMAD7 after transfection with (b) miR-32 agomir and (c) miR-32 antagomir were determined by western blotting. (d) Left runnel: predicted consequential pairing of SMAD7 target region and miR-32 (http://www.targetscan.org); right runnel: luciferase activity of SMAD7 targeting miR-32 after overexpression of miR-32 by transfection with miR-32 agomir in comparison to NC with either wild-type or mutated 3′-UTR of SMAD7 (n = 3/group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
SMAD7 overexpression alleviates fibrosis and EMT induced by miR-32
After we determined the role of miR-32 on fibrosis, EMT, inflammation and autophagy, we further investigated the underlying mechanism. The protein level of SMAD7 by transfection with SMAD7-overexpression plasmid was markedly upregulated compared with parental and empty vector groups (Figure 4(a), ***p ≤ 0.001). Next, SMAD7-overexpression plasmid was cotransfected into HK-2 cells with miR-32 agomir to assess the function of SMAD7. The results of western blot showed that the protein expression level of FN was significantly lower under HG condition in miR-32 agomir + SMAD7 group compared to those in miR-32 agomir + vector group, whereas the levels of E-cadherin and LC3 II/I were greatly increased (Figure 4(b), **p ≤ 0.01, ***p ≤ 0.001).
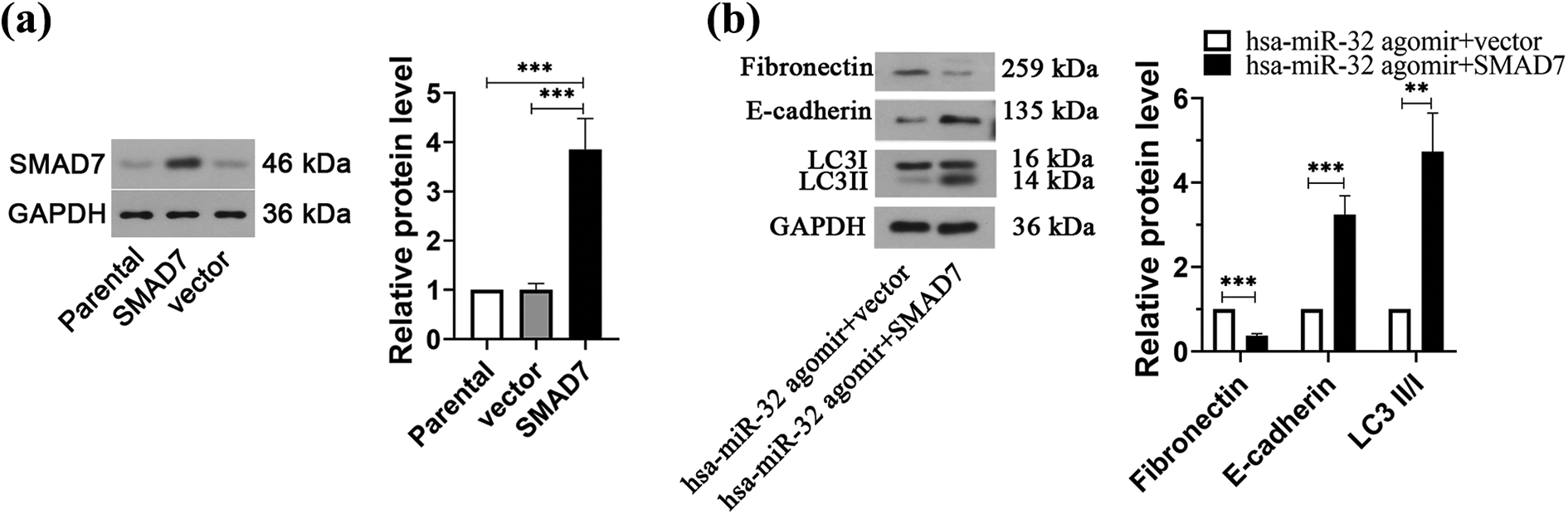
MiR-32 alleviates fibrosis, inflammation and autophagy by the SMAD7 pathway. (a) Western blotting analysis was used to determine the protein level of SMAD7 after transfection with SMAD7-overexpression plasmid. (b) SMAD7-overexpression plasmid was cotransfected into HK-2 cells with miR-32 agomir. The protein levels of FN, E-cadherin and LC3 II/I were detected by western blotting (n = 3/group). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Discussion
There is growing interest in the role of miRs among people in chronic diseases, including diabetic complications. MiRs mediate post-transcriptional regulation of genes through inhibiting target gene mRNA translation. 18,19 In this study, we find miR-32 levels to be greatly upregulated in STZ-induced DN rat kidneys. In line with the findings in DN rats, increased miR-32 was also observed in vitro models of DN. These results show that miR-32 has an unfavorable effect on renal function and might be used to reflect the severity of renal fibrosis in DN. MiR-32 can be efficiently expressed in HK-2 cells by using miR-32 agomir, whereas miR-32 is significantly downregulated after miR-32 antagomir transfection. A previous study has found pharmacological miR-21 silencing can extremely alleviate fibrosis and ameliorate the injury of nephropathy via stimulating metabolic pathways. 8 Consisted with this finding, our study reveals that knockdown of miR-32 significantly decreases the levels of Col IV and FN. Downregulated Col IV and FN expression reduces the deposition of ECM in DN and thereby attenuates fibrosis. 20
EMT serves an important role in the progression of fibrogenesis. During EMT, epithelial cells lose the intrinsic characteristics and induce expression and phenotypic changes of mesenchymal markers thus produce more fibroblast-like cells. 21 Tubular fibroblasts usually are induced to myofibroblasts due to the loss of epithelial marker expression and activation of mesenchymal markers. 22 Several miRs have been reported as fibrosis promoters and underlying prognostic markers in diabetes or diabetes-related complications, such as miR-27a and miR-135a. 23,24 Li et al. found that the pathological silencing of miR-32 could improve hepatic fibrosis. 25 However, there was no direct evidence to support the role of miR-32 in DN fibrosis. In this study, we observe that miR-32 overexpression potentiates EMT process in HG-incubated HK-2 cells, exhibited by the upregulation of α-SMA and vimentin expressions as well as the downregulation of E-cadherin expression. In contrast, a decreased α-SMA and vimentin expression as well as an increased E-cadherin expression are induced by miR-32 silencing. These findings imply that miR-32 silencing may mitigate the degree of EMT in DN. To elucidate the potential molecular mechanisms by which miR-32 involves in the progression of DN, TargetScan and dual-luciferase reporter gene assay are used to confirm SMAD7 as a target gene of miR-32. SMAD7 is a suppressive member of the SMAD family, who participates in the TGFβ/SMAD signaling pathway. 26 A previous study has revealed the protective role of SMAD7 in diabetic kidney disease and overexpression of SMAD7 can efficiently reduce diabetic renal injury. 15 Besides, overexpression of SMAD7 in nephropathy is known to inhibit renal fibrosis. 12 The current study finds that reintroduction of SMAD7 expression rescues miR-32-induced HK-2 cells EMT and renal fibrosis. These results indicate that miR-32/SMAD7 axis can regulate the progress of fibrosis and EMT in DN.
MiR-32 overexpression results in an obvious upregulation of IL-1β and IL-6 levels in HG-treated HK-2 cells. However, silencing of miR-32 inhibits the release of the above inflammatory cytokines, which is in line with a previous study. 27 Our results imply the inhibitory effect of miR-32 silencing on inflammation in DN. In addition, autophagy is a biological regulatory process to maintain metabolic homeostasis. 28 Multiple studies have demonstrated that miRNAs serve as important players in the regulation of autophagy and are involved in DN. 29,30 However, the role of miR-32 on autophagy in DN was unknown. Changes in autophagy are examined by microtubule-associated protein LC3. LC3 is located on the membrane of autophagosomal and is composed of cytosolic LC3 I and membrane-bound LC3 II. In the present study, miR-32 overexpression significantly downregulates LC3 II/I levels and upregulates P62 level, thereby inhibiting autophagy. The conversion of water soluble LC3 I to the autophagosome-associated lipidated form (LC3 II) and the degradation of P62 are observed with the knockdown of miR-32. Combined with the results of LC3 II/I and P62, we think that autophagy is suppressed by miR-32 overexpression in HG-cultured HK-2 cells, whereas knockdown of miR-32 reverses autophagy. These data are in agreement with the results using renal mesangial cells. 17 As miR-32 overexpression inhibits autophagy in HG-cultured HK-2 cells, we speculate that miR-32 may be involved in the regulation of autophagy in DN. A previous report shows that miR-22 promotes renal tubulointerstitial fibrosis and inhibits autophagy by targeting PTEN in DN. 29 As shown in our study, miR-32 overexpression contributes to autophagy suppression and fibrosis in HG-cultured HK-2 cells, whereas overexpression of SMAD7 protectively antagonizes HG- and miR-32-induced autophagy suppression and fibrosis. Therefore, our findings indicate that silencing of miR-32 may induce autophagy and attenuate fibrosis by targeting SMAD7 in DN.
In summary, miR-32 is upregulated in kidneys of DN rats and HG-incubated HK-2 cells. Overexpression of miR-32 inhibits autophagy and promotes fibrosis, EMT and inflammation, whereas the knockdown of miR-32 effectively relieved HG-induced autophagy suppression, fibrosis, EMT and inflammation in HK-2 cells. Reintroduction of SMAD7 expression protectively antagonized HG- and miR-32-induced autophagy suppression, EMT and fibrosis. Although additional exploration is still required, our study indicates that miR-32 may play roles in the process of EMT and fibrosis in DN via targeting SMAD7.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the Natural Science Foundation of Heilongjiang Province (No. LH2020H064).