
Abstract
Silicosis is a lung fibrotic disease caused by chronic silica exposure. Aberrations in long non-coding RNA (lncRNA) expression are associated with fibrotic diseases, but the role of lncRNAs in silicosis pathogenesis remains unclear. Here, we investigated the expression of lncRNAs during silicosis and the role of MRAK050699 in epithelial–mesenchymal transition (EMT). Differentially expressed lncRNAs in the lung tissues of normal and silicosis rats were compared, and their biological effects were determined using the Gene Ontology term and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses. There were 1077 differentially expressed lncRNAs (378 upregulated and 699 downregulated). MRAK052509, MRAK139674, AY539881, MRAK050699, XR_6113, and BC167061 were selected to verify expression in silicosis rats using quantitative reverse transcription polymerase chain reaction. MRAK050699 was knocked down in rat alveolar type II epithelial cells, and the molecular mechanism of transforming growth factor-β (TGF-β)-induced EMT in these cells was studied. All selected lncRNAs were upregulated in the silicosis rats, consistent with the sequencing results. MRAK050699 knockdown inhibited EMT of RLE-6TN cells by regulating the TGF-β/Smad3 signaling pathway. Thus, the differential expression of lncRNAs is related to silicosis development, and MRAK050699 plays an important role in EMT, suggesting a potential therapeutic target for silicosis.
Introduction
Silicosis is a systemic disease caused by long-term exposure to SiO2 dust during its production and is mainly caused by pulmonary diffuse fibrosis. It is considered to be one of the most serious occupational diseases, and there is no specific treatment for it. 1 Pulmonary fibrosis is a typical pathological feature of silicosis. As silica dust stimulation is long term, it can be detected by inflammatory cells and macrophages and can induce the production of transforming growth factor-β (TGF-β). 2 This leads to pulmonary interstitial inflammation, and fibroblast and myofibroblast proliferation, resulting in fibrosis. 3 Therefore, fibroblasts play a significant role in pulmonary fibrosis. 4 Fibroblasts are derived from damaged epithelial cells via a process known as epithelial–mesenchymal transformation (EMT). 5 In this process, epithelial cells lose their phenotypic characteristics and acquire those of mesenchymal cells. Epithelial markers such as E-cadherin are lost, whereas the expression of mesenchymal markers, such as N-cadherin, α-smooth muscle actin, vimentin, and EMT transcription factors (EMT-TFs) ZEB1, ZEB2, and Snail, is increased. 6 Increasing evidence shows that the pathological process of pulmonary fibrosis involves various factors, and the abnormal expression of non-coding RNAs is related to several fibrotic diseases, opening new avenues for research into therapeutic targets for pulmonary fibrosis. 7
Long non-coding RNAs (lncRNAs), which were considered as meaningless “transcriptional noise,” refers to an RNA transcript of length more than 200 bp and lacks an open reading frame and coding potential. With the increasing number of comprehensive studies on lncRNAs in recent years, it can be surmised that they are involved in various pathophysiological processes in several diseases such as pulmonary fibrosis.8,9 For example, recent studies have shown that lncRNA LOC103691771 plays an important role in the differentiation of myofibroblasts induced by TGF-β1, and its abnormal expression is related to the occurrence and development of silicosis. 10 In addition, lncRNAs are competitive endogenous RNAs (ceRNAs), acting as endogenous microRNA (miRNA) “sponges” to regulate the activity of miRNAs. 11 Qian et al. 12 found that the expression of lncRNA ZEB1 antisense RNA 1 (ZEB1-AS1) is upregulated in bleomycin-induced rat lung tissue and TGF-β1-induced rat lung epithelial-T-antigen-negative (RLE-6 TN) cells and positively correlated with the level of ZEB1, the main regulator of EMT. These results suggest that ZEB1-AS1 is a new fibrogenic molecule, which plays a regulatory role in the miR-141-3p/ZEB1 axis during pulmonary fibrosis. Another study also revealed that the expression profiles of 682 lncRNAs are altered (300 upregulated and 382 downregulated) in silica-induced silicosis rat model, indicating that the alteration of lncRNAs occurs in pulmonary fibrosis. 13 These results indicate that lncRNAs are a potential therapeutic target for the prevention and treatment of pulmonary fibrosis.
In this study, we used gene chip technology to study the differential expression of lncRNAs in the lung tissue, bioinformatic tools to analyze the lncRNA–miRNA–mRNA ceRNA network, and in vivo and in vitro experiments to verify the expression profiles. We also explored the role of MRAK050699 in EMT of RLE-6 TN cells induced by silica dust.
Methods
The study was conducted in accordance with the Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies. 14 The animal use protocol was reviewed and approved by the Laboratory Animal Ethical and Welfare Committee of laboratory Animal Center, Ningxia Medical University (Ethics of Ningxia Medical University No. 2020-374).
Animals and treatments
Twenty specific pathogen-free (SPF) male Sprague–Dawley (SD) rats with body weights of 180–220 g were provided by the Experimental Animal Center of Ningxia Medical University. The animal study was reviewed and approved by the Ethics Committee of Ningxia Medical University (Animal License No. SCXK [Ning] 2015-0001) and examined by the Ethics Committee of Ningxia Medical University. After 7 days of adaptive feeding, the rats were randomly divided into two groups: the saline control group (n = 10) and the silicosis model group (n = 10). One milliliter (50 mg/mL) of free SiO2 dust suspension was injected into the trachea of rats in the model group, and the same amount of normal saline (5 mg/kg) was injected into the rats of the control group. The treated rats were then fed normally for 45 days.
Histopathological examination
After 45 days of dust exposure, 10% chloral hydrate (300 mg/kg) was injected intraperitoneally into anesthetized rats. After exposure of the peritoneal cavity through celiotomy, the lung tissue was removed. The upper lobe of the right lung was fixed in 10% paraformaldehyde for 24 h and stained with hematoxylin and eosin (H&E) and Masson’s trichrome. Three sections from each rat were observed in a panoramic field of vision. The pathological changes in the lung tissues in each group were observed at 100× under a light microscope (Leica, Germany). Moreover, the lung tissues of the right lower lobe were immediately frozen in liquid nitrogen and transferred to a −80°C freezer until lncRNA microarray analysis.
Immunohistochemical (IHC) staining
The glass slides with tissue sections were baked in an oven at 65°C for 1 h and dewaxed using xylene solution. After removing the solution, the sections were soaked in 3% H2O2 for 10 min, washed with H2O2, and washed twice in water. Thereafter, citric acid buffer was added on the sections, which were baked for 3 min in the oven and cooled to room temperature. The procedure was repeated once. The slides were then incubated with 0.5% bovine serum albumin for 30 min. The sections were incubated with the primary antibody (proportion of 1:1), at a dilution of 500:1, overnight at 4°C. After phosphate-buffered saline (PBS) flushing, the slides were incubated with the secondary antibody for 30 min and rinsed with PBS. When the color of the sections changed after the addition of 3,3′-diaminobenzidine (DAB) staining solution, the sections were rinsed and re-stained with hematoxylin for 3 min. The sections were then dehydrated by soaking in 75%, 85%, 95%, and 100% alcohol solutions for 5 min each and in xylene for 3 min. This step was repeated twice, and neutral gum was used to seal the slides thereafter. Three sections per rat were observed in a panoramic field of vision.
lncRNA sequencing and bioinformatic analysis
According to the results of the histological analysis, three lung samples were randomly selected from each group for lncRNA expression profile analysis. The rat lncRNA chip is designed to study the expression profile of lncRNAs and protein-coding genes. The data of approximately 9000 lncRNAs were collected from authoritative databases(including the National Center for Biotechnology Information Reference Sequences (NCBI RefSeq) and University of California Santa Cruz all mRNA dataset).
RNA labeling and array hybridization were carried out using the One-Color Microarray-Based Gene Expression Analysis protocol (Agilent Technologies, Inc., Santa Clara, CA, USA) with minor modifications. After the removal of rRNA using the mRNA-ONLY™ Eukaryotic mRNA Isolation Kit (Epicentre Biotechnologies, Madison, WI, USA), mRNA was purified from the total RNA. Each sample was then amplified and transcribed into fluorescent cRNA along the entire length of the transcripts without 3′ bias using a mixture of oligo(dT) and random primers (Arraystar Flash RNA Labeling Kit, Arraystar). The labeled cRNAs were purified using an RNeasy Mini Kit (Qiagen, GmbH, Germany). The concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA) were measured using a NanoDrop ND-1000 UV-Vis Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). The labeled cRNAs were fragmented by mixing 1 μg of each labeled cRNA with 5 μL of 10× blocking agent and 1 μL of 25× fragmentation buffer. The mixture was heated at 60°C for 30 min and diluted by adding 25 μL of 2× GE Hybridization Buffer. Fifty microliters of the hybridization solution was dispensed into a gasket slide and assembled to a microarray slide. The slides were incubated for 17 h at 65°C in an Agilent Hybridization Oven.
The hybridized arrays were washed, fixed, and scanned using the Agilent DNA Microarray Scanner (part number G2505 C). Agilent Feature Extraction software (v11.0.1.1) was used to acquire the chip diagram, read the value, and obtain the original data. Quantile standardization and the subsequent data processing of the original data were carried out using GeneSpringGXv12.1 software (Agilent Technologies). After standardization, high-quality probes were selected (at least 3 of the 6 samples were marked as Present or Marginal) for further analysis. The differentially expressed lncRNAs or mRNAs between the groups were screened by p value/false discovery rate (FDR) based on the fold change (FC). The written script was used for hierarchical clustering.
The target genes of differentially expressed miRNAs were predicted using miRWalk2.0, and the regulatory network of differentially expressed miRNA–target genes was constructed. The miRanda tool was used to predict the binding of the top 100 upregulated and downregulated lncRNAs and miRNAs according to the multiple of difference. The integrated differentially expressed lncRNA–miRNA–mRNA network was constructed using Cytoscape software.
Kyoto Encyclopedia of Genes and Genomes (KEGG) was then used to analyze the enriched pathways of lncRNA-related target genes; Gene Ontology (GO) enrichment analysis was used to analyze the functions of lncRNA-related target genes.
Cell culture and establishment of a co-culture system
RLE-6 TN cells and rat alveolar macrophage NR8383 cells were obtained from subcultures of the American Tissue Culture Collection (Rockville, MD, USA). The cells were cultured in Ham’s F12 medium supplemented with penicillin (final concentration 100 U/mL), streptomycin (final concentration 100 μg/mL), and 10% fetal bovine serum. The experimental groups were as follows: Control group, Silica group, Silica+shRNA-NC group, and Silica+shRNA-MRAK050699 group. Generally, RLE-6 TN cells with or without lentivirus were grown in six-well plates for 24 h until 60%–70% of the cells were fused. At the same time, treated or untreated NR8383 cells were cultured in six-well plates inserted into a transwell chamber. After 24 h of incubation, NR8383 cells were added into the six-well plates of RLE-6 TN cells and grown for another 48 h. In the Control group, uninfected RLE-6 TN cells and untreated NR8383 cells were used. In the Silica group, uninfected RLE-6 TN cells and dust-stimulated NR8383 cells (40 μg/cm2) were used. In the Silica+shRNA-NC group, RLE-6 TN cells infected with shRNA-NC (lentivirus vector transfected with nonsense sequence) and silica-treated NR8383 cells were used. In the Silica+shRNA-MRAK050699 group, RLE-6 TN cells infected with shRNA-MRAK050699 (lentivirus vector transfected with one sequence) and silica-treated NR8383 cells were used.
Quantitative real-time PCR
Primer sequence.

Western blotting
Different concentrations (6%, 8%, and 10%) of sodium dodecyl sulfate-polyacrylamide electrophoresis gels were prepared based on different molecular weights of the proteins to be tested. The total cell protein extract (μg protein/lane) was loaded onto the gel and separated at 80 V for approximately 30 min, and then at 120 V when the samples entered the separation gel to continue electrophoresis. The sealing solution was prepared by dissolving 5% skim milk powder in 1× Tris-HCl buffer, salt solution, and Tween 20 (TBST) solution. The primary antibody was diluted (1:1000) at room temperature for 1 h and incubated with polyvinylidene difluoride membrane overnight at 4°C. The membrane was then washed with TBST three times for 10 min each. For the secondary antibody, sealing was performed at room temperature (23°C–26°C)for 1 h by adding the appropriate concentration of the antibody (1:2500) diluted with gluconate. The membrane was then washed with TBST again. The blot was developed by adding equal volumes of chemiluminescence reagent A solution and liquid B to the membrane and imaged using a chemiluminescence gel imager. Protein expression was analyzed using ImageJ 1.8.0 software, and the relative expression of the proteins was calculated as the gray value of the target protein/the gray value of the internal reference protein.
Immunofluorescence experiment
The lid slides were soaked in 75% alcohol for 2 h and placed in a six-well plate. The cells were inoculated in a six-well plate with approximately 1 × 106 cells/well and cultured at 37°C with 5% CO2. After incubation for 24 h, the upper transwell chamber was removed, and 4% paraformaldehyde was added for 15 min to fix the bottom RLE-6 TN cells. The cells were then rinsed three times, treated with 0.1% Triton X-100, and allowed to rest for 10 min. Sealing was performed for 30 min. Subsequently, the primary antibody was added and incubated overnight at 4°C. After incubation, the slices were washed with PBS solution and incubated with the secondary antibody labeled with fluorescein for 1 h in the dark. The slices were then taken out, washed with PBS three times, placed on the slide, sealed with 4′-diamidino-2-phenylindole (DAPI), and observed under a laser confocal microscope.
Statistical analysis
The experimental data were analyzed using SPSS 22.0 software (SPSS Inc., Chicago, IL, USA) and plotted using GraphPad Prism 7.0 software (CA, USA). All data are expressed as mean ± standard deviation. The gene and protein expression levels among the groups were compared using one-way ANOVA test. Pairwise comparisons between groups were performed using the Student–Newman–Keuls method. The data were tested for normality (Shapiro–Wilk test) and variance uniformity (Bartlett’s test). We used t-test to compare the difference in body weight between the control group and the silicosis group. The test level was α = 0.05.
Results
Histopathological evaluation of rat lung tissues
The anatomical dissection revealed that the lungs of rats in the control group were pink, smooth, soft, and elastic, whereas those in the model group had scattered gray-white hard plaques. H&E staining showed that the alveolar structure of rats in the control group was relatively clear, but the blood vessels were slightly dilated and congested. In contrast, the alveolar structure in the model group was destroyed, and nodular structure, emphysema, and some patchy fibrosis were present (Figure 1(a)). Masson staining showed that there was no proliferation of collagen fibers in the alveolar septum of rats in the control group; conversely, blue-dyed collagen distribution was observed in the model group, indicating collagen proliferation in the nodules (Figure 1(a)). IHC staining showed that COL I and α-SMA were expressed at higher levels in the model group than in the control group and were found in interstitial fibrosis and silicotic nodules (Figure 1(b)). These results showed that the rat silicosis model was established successfully. Establishment of a rat model of silicosis. (a) In the rats exposed to silica for 45 days, H&E and Masson staining showed that the silicosis rats had pulmonary fibrosis after being exposed to dust. (b) Immunohistochemical staining of COLI and α-SMA showed that their expression was significantly increased in the silicosis group. H&E: hematoxylin & eosin.
Differential expression of lncRNAs in silica-induced pulmonary fibrosis in rats
As shown in Figure 2, 1077 lncRNAs were differentially expressed at FDR <0.05. Among them, 378 were upregulated and 699 were downregulated. Among mRNAs, 3452 were differentially expressed; 1283 were upregulated and 2169 were downregulated. Among the differentially expressed miRNAs with p < 0.05, 14 were upregulated and 25 were downregulated. Cluster analysis of mRNA expression profile and expression of fibrosis-specific lncRNAs in silicosis. lncRNA: long non-coding RNA.
Biological functions of differentially expressed lncRNAs
The biological functions of dysregulated lncRNAs in silicosis were determined by GO term and KEGG pathway enrichment analyses. The pathway enrichment analysis showed that the upregulated lncRNAs were mostly enriched in complement and coagulation cascades, starch and sucrose metabolism, rheumatoid arthritis, pentose and glucuronate interconversions, porphyrin and chlorophyll metabolism, pertussis, ascorbate and aldarate metabolism, Staphylococcus aureus infection, steroid hormone biosynthesis, and Toll-like receptor signaling pathway (Figure 3(a)). Moreover, the pathways that were significantly related to the downregulated lncRNAs were the Rap1 signaling pathway, endocytosis, MAPK signaling pathway, proteoglycans in cancer, regulation of actin cytoskeleton, T cell receptor signaling pathway, natural killer cell-mediated cytotoxicity, Ras signaling pathway, cancer pathways of miRNAs, and PI3K-Akt signaling pathway (Figure 3(b)). Biological functions of differentially expressed lncRNAs. (a, b) KEGG pathway of differentially expressed lncRNAs. (c–e) GO term enrichment analysis of highly expressed lncRNAs. lncRNA: long non-coding RNA, KEGG: Kyoto Encyclopedia of Genes and Genomes, GO: Gene Ontology.
In the GO term enrichment analysis, we found that the highly expressed lncRNA-related target genes were enriched in 72 functional items in the cell components, and the top 10 terms are shown in Figure 3(c). Their molecular functions were mostly enriched in 145 functional items, which are shown in Figure 3(d). The top 10 terms are shown in the figure, and 997 functional items were enriched in the biological process as shown in Figure 3(e). These indices may be related to pulmonary fibrosis in rats. These terms are primarily related to the extracellular region, response to external stimulus, and transporter activity.
Verification of differentially expressed genes
We sorted the lncRNAs according to their FC values and randomly selected those with an FC value greater than 5. RT-qPCR was then performed to determine their change level. In the silicosis samples, MRAK052509, MRAK139674, AY539881, MRAK050699, XR_6113, and BC167061 were selected for verification, and the results showed that their expression was significantly increased compared to the normal rat lung tissue samples (Figure 4), consistent with the results of gene sequencing. RT-qPCR verification of differentially expressed lncRNAs in rats with silica-induced pulmonary fibrosis. lncRNA: long non-coding RNA, RT-qPCR: quantitative reverse transcription polymerase chain reaction.
Morphological changes in dust-stimulated NR8383 cells
NR8383 cells were stimulated with 40 μg/cm SiO2 dust, and the morphological changes in RLE-6 TN cells were observed. The cells in the control group showed typical epithelioid characteristics, were polygonal and oval, and had tight junctions (Figure 5(a)). Cells in the silicosis group showed interstitial-like morphology, with long spindles and wide intercellular spaces (Figure 5(b)). Similarly, the knockdown control group showed interstitial-like cell morphology, but the shape changed from oval to spindle, and the cells had tight junctions (Figure 5(c)). Only a small proportion of cells in the knockdown group changed from oval to a long shuttle or spindle shape, and the cells were tightly connected (Figure 5(d)). Morphology of RLE-6TN cells (×100). NR8383 cells were stimulated with 40 μg cm−2 SiO2 dust, and the morphological changes in RLE-6TN cells were observed under a microscope. A: normal group; B: model group; C: knockout control group; D: knockout group.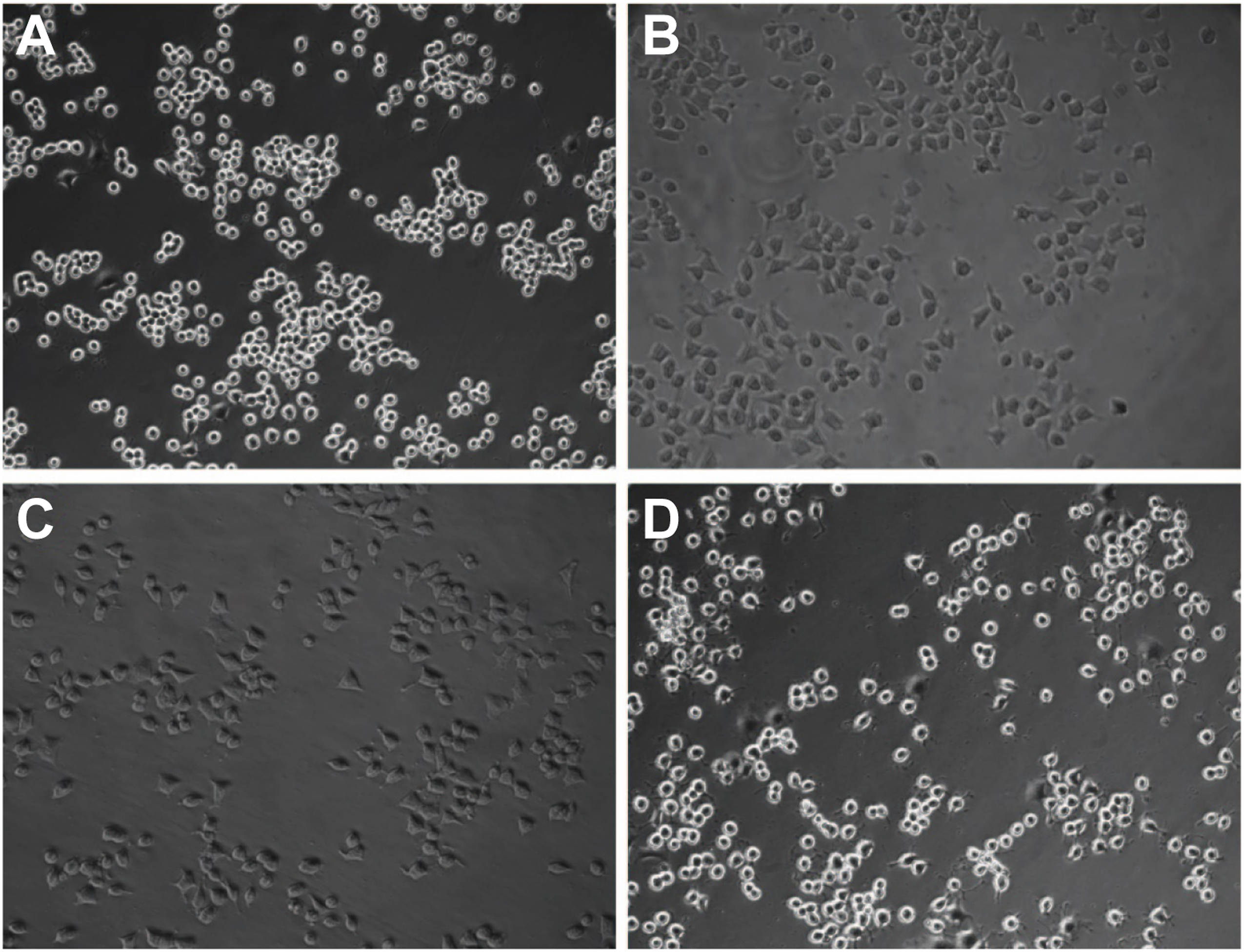
Effect of MRAK050699 on EMT of RLE-6TN cells
The low expression vector of MRAK050699 was transfected into RLE-6TN cells to form a stable MRAK050699 knockdown cell line. The western blot images are shown in Figure 6(a). NR8383 cells treated with silica and co-cultured with RLE-6TN cells presented significantly reduced levels of the epithelial cell-related protein E-cadherin (Figure 6(b)) and increased levels of the interstitial cell-related proteins N-cadherin, α-SMA, and vimentin in RLE-6TN cells compared to the untreated cells (Figures 6(c)-(e)). Compared with the shRNA-NC group, RLE-6TN cells with knockdown of the MRAK050699 gene and co-cultured with dust-stimulated NR8383 cells showed significantly increased expression of E-cadherin (Figure 6(b)) and decreased expression of N-cadherin, α-SMA, and vimentin in RLE-6TN cells (Figures 6(c)-(e)). These results are consistent with those of RT-qPCR (Figure 7(a)). Western blotting of fibrosis factors in different experimental groups. (a) Western blotting of the proteins in: Lane 1: RLE-6TN cells, Lane 2: silica-treated cells, Lane 3: knockdown control, and Lane 4: MRAK050699 knockdown cells. Graphs showing the relative expression of (b) E-cadherin, (c) N-cadherin, (d) vimentin, and (e) α-SMA in the assay. RT-qPCR analysis of related genes in different experimental groups. The relative expression levels of (a) TGFBR1, P-Smad3, E-cadherin, N-cadherin, α-SMA, and vimentin; (b) Snail, ZEB1, and ZEB2; and (c) COLI, COLIII, and FN are shown. RT-qPCR: quantitative reverse transcription polymerase chain reaction.

RT-qPCR was then used to detect the expression of the related genes. The expression of fibrosis factors COLI, COLIII, and FN increased in RLE-6TN cells co-cultured with dust-stimulated NR8383 cells. Moreover, in the transfected cells, the expression of the above fibrosis factors in RLE-6TN cells with MRAK050699 knocked down was lower than that in the group transfected with the empty virus (Figure 7(c)).
The expression of some EMT-induced transcription factors (EMT-TFs), such as Snail, ZEB1, and ZEB2, also changed significantly among the groups (Figure 7(b)). Dust-treated NR8383 cells co-cultured with RLE-6TN cells showed increased expression of Snail, ZEB1, and ZEB2 in RLE-6TN cells (Figure 7(b)). When MRAK050699 was knocked down in RLE-6TN cells co-cultured with silica-treated NR8383 cells, the expression of Snail, ZEB1, and ZEB2 in RLE-6TN cells significantly decreased compared with that in the shRNA-NC group (Figure 7(b)).
Immunofluorescence revealed that the knockdown of MRAK050699 in RLE-6TN cells inhibited silica-induced EMT. The expression of TGFBR1, fibronectin, COLI, and COLIII in RLE-6TN cells was significantly reduced in the MRAK050699 knockdown group compared to that in the control group (Figures 8(a)-(h)). A similar trend was observed for vimentin, α-SMA, and N-cadherin in RLE-6TN cells with MRAK050699 silencing. Conversely, silica treatment of RLE-6TN cells resulted in an increased expression of these proteins. Only E-cadherin expression was significantly increased in knockdown cells (Figure 9). This is consistent with the results of western blotting (Figure 6) and RT-qPCR (Figure 7). Immunofluorescence staining of (a, b) TGFBR1, (c, d) fibronectin, (e, f) COL I, and (g, h) COL III in control, silica-treated, knockdown control, and MRAK050699 knockdown cells. The nuclei were counterstained with DAPI (blue: In the left column of the composite drawing), and the antibodies used for the proteins were labeled with nestin (red: In the middle column of the combined drawing). DAPI: 4′-diamidino-2-phenylindole. Immunofluorescence staining of (a, b) E-cadherin, (c, d) vimentin, (e, f) α-SMA, and (g, h) N-cadherin in knockdown cells. The nuclei were counterstained with DAPI (blue: In the left column of the composite drawing), and the antibodies used for the proteins were labeled with nestin (red: In the middle column of the combined drawing). DAPI: 4′-diamidino-2-phenylindole.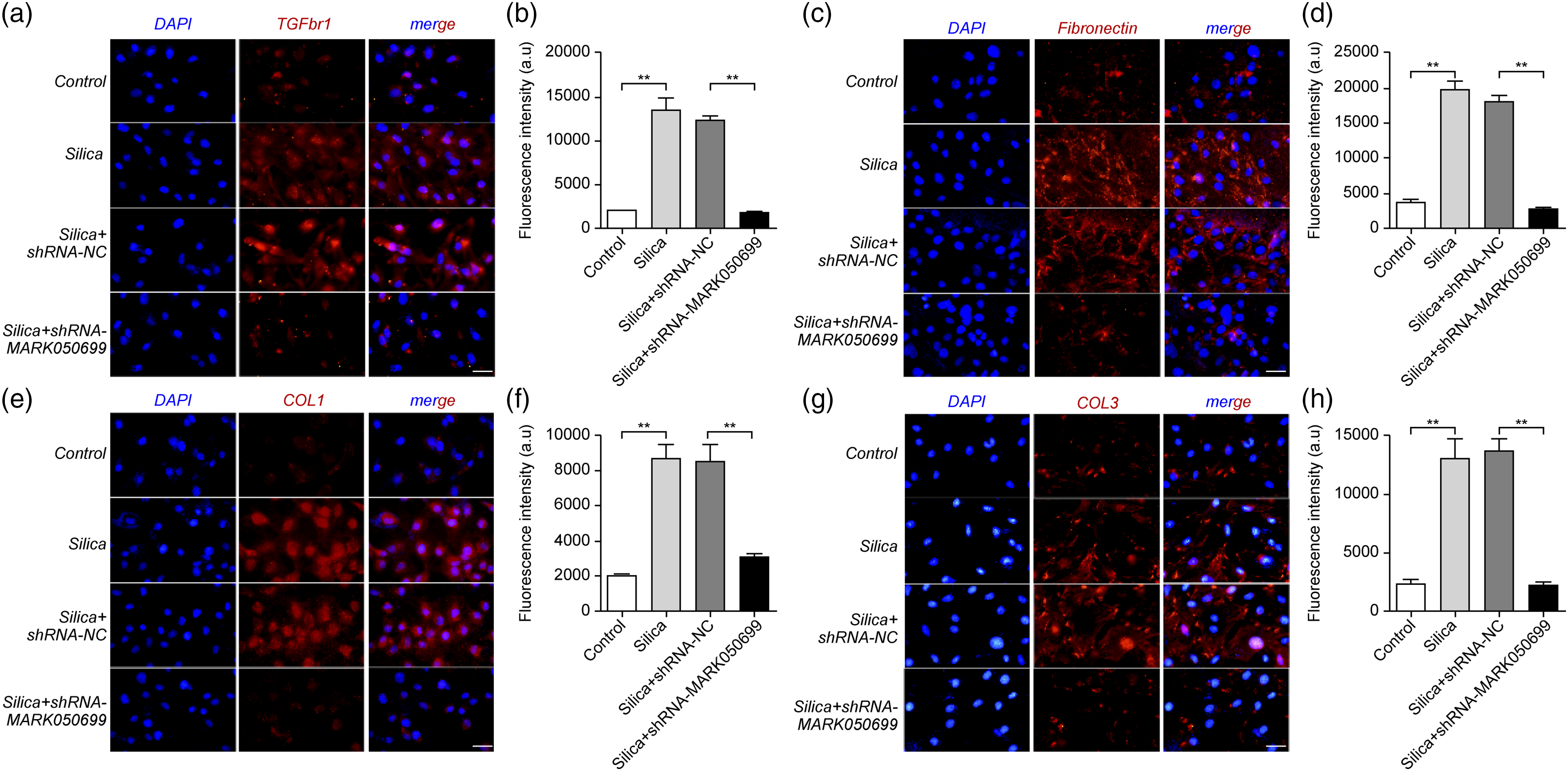

Discussion
Silicosis, which is mainly caused by long-term exposure to silica dust, is one of the most serious occupational diseases in China. Alveolar macrophages play an important role in the pathogenesis of silicosis. 15 After phagocytosis of silica particles, alveolar macrophages stimulate the production of reactive oxygen species, chemokines, and cytokines, excessive proliferation of lung fibroblasts, and excessive deposition of collagen and extracellular matrix. 16 This leads to the formation of silicotic nodules and development of pulmonary fibrosis. Therefore, we used silica to stimulate rat alveolar macrophage NR8383 cells, which were then co-cultured with rat alveolar type II epithelial cells to establish a real-time in vitro silicosis model that can better simulate the cellular environment of silicosis in vivo.
According to recent reports, lncRNAs affect the EMT process in various diseases such as breast cancer, 17 liver cancer, 18 and lung cancer, 19 and fibrosis is the pathological process that precedes or follows cancer development. Therefore, the role of lncRNA in pulmonary fibrosis can be investigated to obtain a new target for treatment. When macrophages are continuously stimulated by silica, the fibrogenic factor TGF-β is released, inducing Smads to activate EMT-TFs, which increase the expression of TGF-β ligand and make cells maintain their EMT state. Thus, lncRNAs can regulate EMT through the TGF-β/Smads signaling pathway.20,21 In addition, one of the central targets of these transcriptional regulators, such as Snail, ZEB1, and ZEB2, is the E-cadherin gene, and its expression is inhibited by these molecules (20,21). The level of E-cadherin is limited, at the same time, N-cadherin expression levels increased. 22
The silicosis rat model was established to detect the changes in the expression profiles of lncRNAs in rats exposed to silica. The results showed that there were 1077 differentially expressed lncRNAs, 378 of which were upregulated and 699 were downregulated. H&E staining showed that there were silicotic nodules in the silicosis model group. IHC staining and western blotting showed that the expression of type I collagen and α-SMA in the silicosis model group was significantly higher than that in the control group. These proteins are associated with fibrosis and are sometimes used as markers of fibrosis, which demonstrates that the silicosis model was established successfully.
The GO term enrichment analysis revealed that the function of differentially expressed lncRNAs in silicosis was mostly related to extracellular region, response to external stimulus, and transporter activity. In KEGG pathway analysis, differentially expressed lncRNAs were found to participate in complement and coagulation cascades, starch and sucrose metabolism, rheumatoid arthritis, Rap1 signaling pathway, endocytosis, and MAPK signaling pathway. Therefore, the differential expression of lncRNAs in these processes may be involved in the pathogenesis of silicosis.
To verify the sequencing results, we selected six differentially expressed lncRNAs and checked their expression in silicosis samples using RT-qPCR. The results showed that the expression of MRAK052509, MRAK139674, AY539881, MRAK050699, XR_6113, and BC167061 in the silicosis group was significantly higher than that in the control group, which was similar to the results of sequencing.
Macrophages exposed to silica secrete effector factors and act on alveolar type II epithelial cells, resulting in EMT. The cells in the normal group showed typical epithelioid characteristics, whereas those in the model group were interstitial-like. Immunofluorescence and western blotting showed that silica could induce EMT in vivo and in vitro. Compared with the group transfected with an empty virus, the knockdown of MRAK050699 in RLE-6TN cells increased the expression of epithelial genes, decreased the expression of interstitial genes, and decreased the expression of some related transcription factors. These results suggest that MRAK050699 promotes EMT in RLE-6TN cells by regulating the TGF-β/Smad pathway.
In conclusion, the differential expression of lncRNAs may be related to the occurrence and development of silicosis, and MRAK050699 may be a potential therapeutic target for silicosis.
Footnotes
Ethical approval
The animal use protocol was reviewed and approved by the Laboratory Animal Ethical and Welfare Committee of laboratory Animal Center, Ningxia Medical University (approval number 2016-128).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported by the National Natural Science Foundation of China (NO.82060584;81660534); Ningxia Natural Science Foundation(2020AAC02019);Excellent Youth Foundation project of Ningxia Education Department(NGY2020035)