
Abstract
Acute kidney injury (AKI) is an independent risk factor for chronic kidney disease (CKD). However, the role and mechanism of microRNA (miRNA, miR) in AKI-CKD transition are elusive. In this study, a murine model of renal ischemia/reperfusion was established to investigate the repairing effect and mechanism of miR-101a-3p on renal injury. The pathological damage of renal tissue was observed by hematoxylin and eosin and Masson staining. The levels of miR-101, profibrotic cytokines, and epithelial–mesenchymal transition (EMT) markers were analyzed using Western blotting, real-time polymerase chain reaction, and/or immunofluorescence. MiR-101 overexpression caused the downregulation of α-smooth muscle actin, collagen-1, and vimentin, as well as upregulation of E-cadherin, thereby alleviating the degree of renal tissue damage. MiR-101 overexpression mitigated hypoxic HK-2 cell damage. Collagen, type X, alpha 1 and transforming growth factor β receptor 1 levels were downregulated in hypoxic cells transfected with miR-101 mimic. Our study indicates that miR-101 is an anti-EMT miRNA, which provides a novel therapeutic strategy for AKI-CKD transition.
Introduction
The prevalence of acute kidney injury (AKI) and chronic kidney disease (CKD) has been increasing over time. In the past few years, AKI was an independent risk factor for CKD. 1 A large amount of data from experimental animals and humans have been published, indicating that AKI can lead to incomplete repair of the kidney and eventually result in tubulointerstitial fibrosis, which is a hallmark of chronic progressive kidney disease. This phenomenon is called AKI-CKD transition. 2 However, AKI-CKD progression is a complicated process that is still not fully understood. 3
MicroRNAs (miRNAs, miRs) are small endogenous non-coding RNAs of approximately 18–22 nucleotides. MiRNA plays a vital role in homeostasis of normal human bodies and various biological processes of cells. 4 The levels of miR-101 have been reported to increase significantly in the serum of AKI patients. 5 MiR-101 overexpression alleviated the degree of AKI via inhibiting the release of IL-2 from CD4+ T cells in peripheral blood of AKI patients. 6 In addition, the role of miR-101 in other fibrotic disease models has been reported. MiR-101 suppressed liver fibrosis by downregulating the PI3K/AKT/mTOR pathway. 7 Another study indicated that miR-101 restrained fibroblast proliferation and activation, thereby attenuating pulmonary fibrosis. 8 MiR-101 also inhibited myocardial fibrosis by targeting c-Fos and transforming growth factor β receptor 1 (TGFβR1) pathway. 9 However, the expression levels and roles of miR-101 are poorly understood in AKI-CKD progression. Based on the above background, we speculate that miR-101 overexpression can suppress renal fibrosis and slow the progression of AKI-CKD.
Previous evidence has shown that epithelial–mesenchymal transition (EMT) plays an important role in renal fibrosis. 10 Inhibition of EMT could effectively alleviate renal fibrosis and suppress the progression of AKI-CKD. 11 A study found that the loss of miR-101 accelerated EMT process in hepatocytes. 12 Another research reported that miR-101 overexpression inhibited the processes of EMT and migration in ovarian cancer cells by modulating ZEB1 expression. 13 Therefore, we hypothesize that miR-101 plays an important role in AKI-CKD transformation by regulating EMT process.
To prove this hypothesis, we used a murine model of ischemia/reperfusion (I/R) injury and hypoxia-induced human kidney proximal tubular epithelial (HK-2) cells to explore the role and mechanism of miR-101a-3p in AKI-CKD transition.
Methods
Animals
Twenty-four C57BL/6 male mice aged 6–8 weeks were obtained from Liaoning Changsheng Biotechnology Co., Ltd (Benxi, China). All animals had free access to food and water. The handling of experimental animals followed the use and care guidelines of the Ethical Committee of Animal Experiments. Informed consent was obtained from all individual participants included in the study.
Renal I/R injury
Mouse models of I/R injury were established as previously described. 2 Briefly, the mice were anesthetized with pentobarbital sodium. The ischemia was performed on a 38°C heating pad for 35 min by clamping of left renal hilus, and then the left kidney was reperfused by releasing the clamp. Contralateral kidneys were removed 35 days after I/R injury and mice were killed 42 days after surgery. Left kidneys and peripheral blood were collected. The experiment of sham-operated control mice was performed without clamping of left renal hilus (n = 6).
Cell culture
HK-2 and 293T cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HK-2 and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, 41500-067; Gibco, Carlsbad, California, USA) containing 10% fetal bovine serum (SH30084.03; Hyclone, Logan, Utah, USA), respectively. For hypoxic culture, cells were placed in a hypoxic incubator (Thermo, Waltham, Massachusetts, USA) at 37°C with 1% O2 and 5% CO2 for 48 h. Control cells were incubated for 48 h under normoxic conditions (21% O2, 5% CO2, 37°C).
Vector constructions and cell transfection
The mice miR-101-overexpressed lentiviral vector (mmu-miR-101), human miR-101 (hsa-miR-101) mimics and their negative control were obtained from GenePharma (Shanghai, China). Delivery of lentivirus in vivo was conducted as described earlier. 14 Briefly, the mmu-miR-101-overexpressed lentivirus was introduced into mice by intravenous injection (1 × 109 TU/mL, 150 µL per mouse) at 24 h after I/R injury, and an empty lentivirus vector was used as a negative control. Mice were euthanized at 42 days after I/R injury. Cell transfection in vitro was performed following the previous literature. 15 In brief, HK-2 cells were transfected with hsa-miR-101 using a Lipofectamine 2000 Kit (11668-019; Invitrogen, Carlsbad, California, USA), according to the manufacturer’s protocols. Transfected cells were collected 48 h after incubation.
Histology
The renal tissues were fixed with 4% (w/v) paraformaldehyde for 24 h, and paraffin-embedded renal tissues from mice were cut into 5 µm sections. Kidneys sections were stained using hematoxylin and eosin (HE) staining and Masson staining methods. The fibrotic area was observed by light microscopy (Nikon, Tokyo, Japan) at original magnification 200×.
Immunofluorescence
Immunofluorescence was conducted as previously described. 3 Briefly, 5 µm tissue slides were incubated with antigen retrieval solution (9 mL of 0.1 M citric acid, 41 mL of 0.1 M sodium citrate were mixed with 450 mL distilled water) for 10 min, and blocked in goat serum for 15 min at room temperature (RT). The slides were subsequently incubated with primary antibodies including α-smooth muscle actin (α-SMA, 1:100 dilution in phosphate buffered solution, bs-0189 R; Bioss, Beijing, China), type 1 collagen (Collagen-1, 1:100, ab34710; Abcam, Cambridge, UK), E-cadherin (1:100, #14472; CST, Boston, Massachusetts, USA), vimentin (1:100, #5741; CST) overnight at 4°C, respectively. The sections were incubated with Cy3-conjugated goat anti-rabbit IgG antibody (1:200, A0516; Beyotime, Shanghai, China) as the secondary antibody at RT for 1 h. Nuclear staining was carried out using 4′,6-diamidino-2-phenylindole (C1002; Beyotime) at RT for 5 min. The immunofluorescence was observed by light microscopy (Nikon) at original magnification 400×.
Blood urea nitrogen and serum creatinine assay
For evaluating renal function, peripheral blood samples were collected 35 days after I/R and serums were obtained by centrifugation at 10,000 g (H-2050R; Xiangyi, Hunan, China) for 5 min at 4°C. The contents of blood urea nitrogen (BUN) and serum creatinine (Scr) were determined using BUN or Scr kit (C013-2/C011-1; Jiancheng, Nanjing, China) based on the manufacturer’s protocols.
Enzyme-linked immunosorbent assay
The serum level of neutrophil gelatinase-associated lipocalin (NGAL) at 42 days after I/R injury was measured using an enzyme-linked immunosorbent assay kit (SEB388Mu; Uscnk, Wuhan, China) following the manufacturer’s instruction.
Western blotting
The renal tissues or cells were lysed on ice for 5 min and supernatants were collected after centrifugation (10,000 g, 5 min, 4°C). The bicinchoninic acid (BCA) method was used for the quantification of cellular lysates. A measure of 20 µL of supernatants was fractionated on 8–10% sodium dodecyl sulfate-polyacrylamide gels before transferring onto polyvinylidene fluoride membranes (IPVH00010; Millipore, Billerica, Massachusetts, USA). Membranes were blocked with 5% (m/v) skim milk (A600669; Sangon Biotech, Shanghai, China) at RT for 1 h and incubated overnight at 4°C via using the following primary antibodies: NGAL (1:1000, A2092; ABclonal, Wuhan, China), α-SMA (1:500, 55135-1-AP; Proteintech, Beijing, China), TGFβR1 (1:1000, A0708; ABclonal), collagen, type X, alpha 1 (COL10A1, 1:500, A13288; ABclonal), E-cadherin (1:500, #14472; CST), vimentin (1:500, #5741; CST), and glyceraldehyde-3-phoshate dehydrogenase (GAPDH, 1:10,000, 60004-1-Ig; Proteintech). The goat anti-rabbit or anti-mouse IgG antibody (1:3000, SE134/SE131; Solarbio, Beijing, China) was used as secondary antibody for 1 h at 37°C. GAPDH was applied as an internal control. Immunoreactive proteins were then visualized by employing the electrogenerated chemiluminescence (ECL) system (PE0010; Solarbio).
Quantitative real-time polymerase chain reaction
Total RNA was isolated from renal tissues and cells using Total RNA Isolating Kit (DP419; Tiangen, Beijing, China) according to the manufacturer’s instructions, and then the total RNA was reverse transcribed into complementary DNA using an RT Primer (Genscript, Nanjing, China) and RNase inhibitor (DP418; Tiangen). Real-time polymerase chain reactions (RT-PCRs) were carried out with the SYBR Green PCR Kit (SY1020; Solarbio). After PCR, the 2−ΔΔCt comparative method was used to calculate the Ct for the relative levels of mRNA and miR-101. The miRNA level was normalized to that of U6, and GAPDH was used as an internal control of mRNA level. The specific primer sequences are presented in Table 1.
Primers for quantitative RT-PCR.

RT-PCR: real-time polymerase chain reaction; α-SMA: α-smooth muscle actin; COL10A1: collagen, type X, alpha 1; TGFβR1: transforming growth factor β receptor 1; miR: microRNA.
Dual-luciferase reporter gene assay
293T cells were inoculated on 12-well plates and co-transfected with pmirGLO-miR-101 and luciferase reporter plasmid using Lipofectamine 2000 (11668-019; Invitrogen) in DMEM without serum. Cells were collected after 48 h and their luciferase activity was measured with Dual-Luciferase Reporter Assay Kit (KGAF040; KeyGene, Wageningen, Netherlands) according to the manufacturer’s instructions. Renilla luciferase activity was used as internal control. The results were expressed by the ratio of the luciferase activity and the renilla luciferase activity.
Statistical analysis
Data are analyzed as the mean ± standard deviation and all experiments in vitro are performed in triplicate. The two-tailed unpaired Student’s t-test and one-way analysis of variance were used to compare the differences of data. Statistical analysis was carried out using GraphPad Prism 8.0. A value of p < 0.05 was considered statistically significant. All the experiments including kidney pathological assessment and statistical processes were completed by at least two professional investigators, who were blind to this study.
Results
MiR-101 is downregulated in AKI-CKD transition
The renal morphology of mice at 42 days after I/R was analyzed by HE staining and Masson’s trichrome staining methods. The results of HE staining showed that the kidney tissues of the mice appeared inflammatory infiltration and obviously vascular proliferation 42 days after I/R injury, compared with the sham-operated control. Part of the glomerular structure was destroyed. Masson staining revealed that I/R injury induced collagen deposition in tubulointerstitium and glomerulus (Figure 1(a)). Besides, the mmu-miR-101 expression level was decreased in the mice kidney tissues in AKI-CKD transition (Figure 1(b)). Figure 1(c) revealed that the levels of BUN and Scr were increased after removal of contralateral kidneys 7 days before measurements. The AKI-CKD transition marker NGAL was overexpressed at 42 days after I/R injury, and miR-101 expression significantly decreased the level of NGAL (Online Supplemental Figure S1(a)).

MiR-101 is downregulated in AKI-CKD transition. (a) Representative photomicrographs of HE and Masson’s trichrome-stained kidney sections are shown at 42 days after I/R injury (bars = 100 μm). (b) As analyzed by quantitative RT-PCR, miR-101 expression is observed at 42 days after I/R injury. (c) Renal function is assessed by measuring BUN and Scr at 42 days after I/R. Data are presented as means ± SD, **p < 0.01, ***p < 0.001; n = 6. AKI: acute kidney injury; CKD: chronic kidney disease; I/R: ischemia/reperfusion; BUN: blood urea nitrogen; Scr: serum creatinine; HE: hematoxylin and eosin; miR: microRNA; RT-PCR: real-time polymerase chain reaction; SD: standard deviation.
Overexpression of miR-101 attenuates renal fibrosis in AKI-CKD transition
To further explore the mechanistic link between miR-101 and AKI-CKD transition, these mice with I/R injury were challenged using miR-101 overexpression lentivirus. These mice exhibited robust miR-101 overexpression as indicated by quantitative RT-PCR (Figure 2(a)). Renal histological sections revealed the overexpression of miR-101 attenuated glomerulosclerosis and interstitial fibrosis (Figure 2(b)). Kidney tissues at 42 days after I/R injury displayed renal interstitial fibrosis and fibroblast accumulation (Figure 2(c)) as showed by the quantification of collagen-1-positive area and α-SMA-positive area (Figure 2(d)), whereas miR-101 overexpression effectively attenuated kidney fibrogenesis and AKI-CKD progression (Figure 2(c) and (d)). Reduced fibroblast accumulation was correlated with decreased mRNA and protein expression levels of α-SMA (Figure 2(a) and (e)). The improvement of tubulointerstitial fibrosis in AKI-CKD progression was associated with overexpression of miR-101 and loss of α-SMA mRNA expression level.

Overexpression of miR-101 attenuates renal fibrosis in AKI-CKD transition. (a) RT-PCR is used to analyze mRNA expression levels of miR-101 and α-SMA. (b) Representative photomicrographs of HE and Masson’s trichrome-stained kidney sections are shown (bars = 100 μm). (c) Sections immunolabeled with primary antibodies against collagen-1 and α-SMA (bars = 50 μm) are shown. (d) Relative areas positive for collagen-1 and α-SMA are quantified. (e) α-SMA protein expression level is analyzed by Western blotting. Data are presented as means ± SD, **p < 0.01, ***p < 0.001; n = 6. AKI: acute kidney injury; CKD: chronic kidney disease; miR/mRNA: microRNA; RT-PCR: real-time polymerase chain reaction; α-SMA: α-smooth muscle actin; HE: hematoxylin and eosin; SD: standard deviation.
Overexpression of miR-101 suppresses EMT process in AKI-CKD transition
The miR-101 overexpression lentivirus was transduced into mice kidney tissues to determine the effects of miR-101 on EMT of AKI-CKD progression. E-cadherin and vimentin were used to quantitatively assess the EMT. RT-PCR and Western blotting revealed that miR-101 overexpression led to a significant increase in mRNA and protein expression levels of E-cadherin, while the expression levels of vimentin were reduced (Figure 3(a) and (b)). Besides, overexpression of miR-101 downregulated the mRNA and protein expression levels of COL10A1 and TGFβR1 in renal tissues as indicated by RT-PCR and Western blotting (Figure 3(c) and (d)).
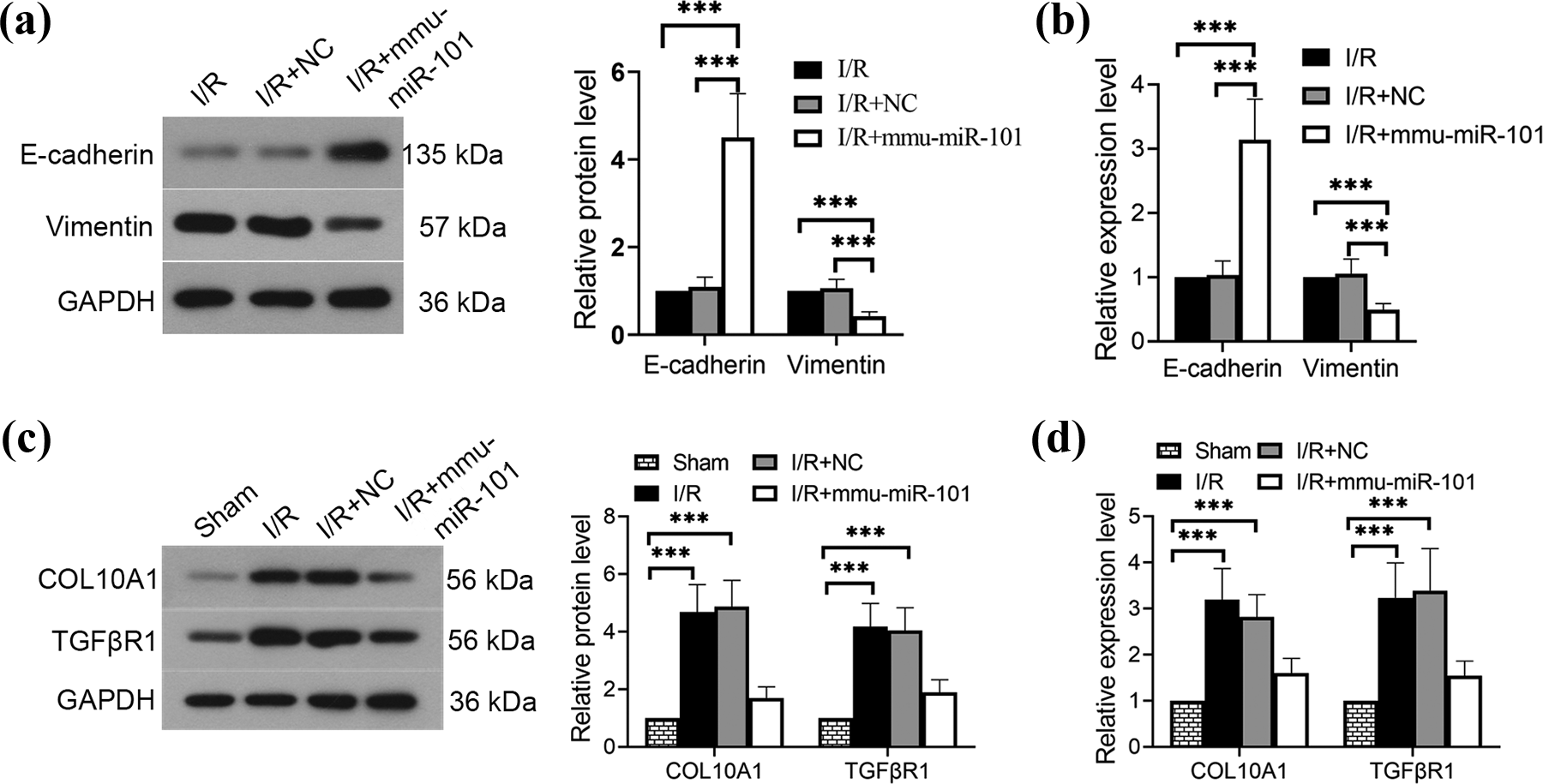
Overexpression of miR-101 suppresses EMT in AKI-CKD progression. The expression of EMT relevant markers (a) E-cadherin, (b) vimentin, (c) COL10A1, and (d) TGFβR1 at 42 days after I/R injury are determined by Western blotting and quantitative RT-PCR, respectively. Data are presented as means ± SD, ***p < 0.001; n = 6. EMT: epithelial–mesenchymal transition; AKI: acute kidney injury; CKD: chronic kidney disease; miR: microRNA; I/R: ischemia/reperfusion; RT-PCR: real-time polymerase chain reaction; SD: standard deviation; COL10A1: collagen, type X, alpha 1; TGFβR1: transforming growth factor β receptor 1.
Overexpression of miR-101 inhibits EMT process in hypoxic HK-2 cells
HK-2 cells incubated under hypoxic conditions (1% O2 and 5% CO2) downregulated expression of E-cadherin and upregulated expression of vimentin and α-SMA, compared with those in normoxic HK-2 cells (Figure 4(a)). Besides, miR-101 expression led to an increase of the E-cadherin expression and a reduction of vimentin and α-SMA expression in hypoxic HK-2 cells (Figure 4(a)). RT-PCR showed that miR-101 expression upregulated the expression level of hsa-miR-101 under hypoxic conditions (Figure 4(b)). Immunofluorescence staining confirmed that the expression of hsa-miR-101 led to the increase of E-cadherin and the decline of vimentin in hypoxic HK-2 cells (Figure 4(c)), thereby suppressing EMT process in hypoxic HK-2 cells. Further study showed that miR-101 overexpression reversed the high expression of NGAL in hypoxic HK-2 cells (Online Supplemental Figure S1(b)).
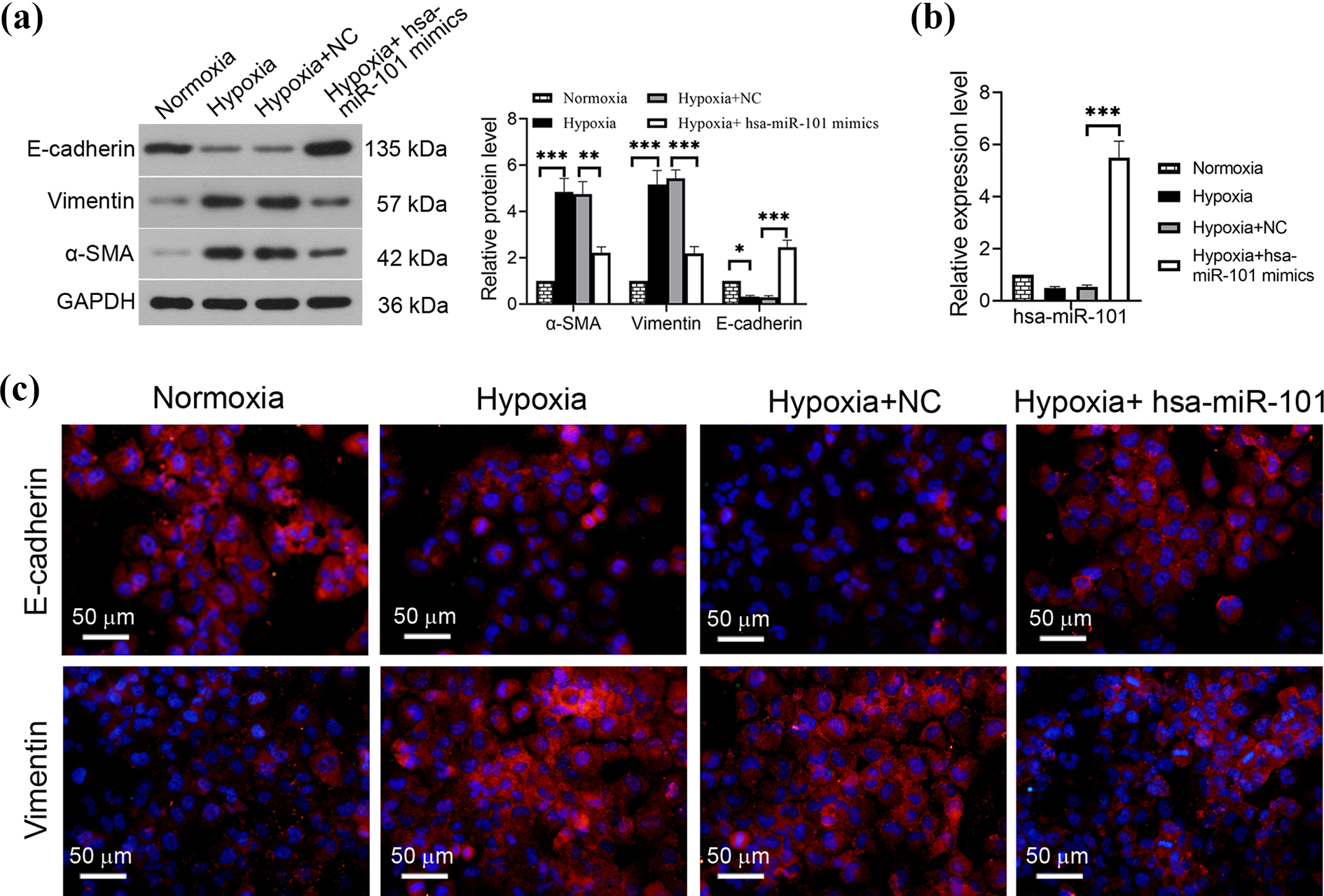
Overexpression of miR-101 inhibits EMT process in hypoxic HK-2 cells. (a) Western blotting is used to detect the protein expression levels of epithelial marker (E-cadherin) and mesenchymal markers (vimentin and α-SMA) in HK-2 cells under normoxic or hypoxic conditions. (b) The expression of miR-101 in HK-2 cells is determined by quantitative RT-PCR. (c) EMT phenotype changes of HK-2 cells under hypoxia are further confirmed with immunofluorescence analysis (bars = 50 μm). Data are presented as means ± SD, *p < 0.05, **p < 0.01, ***p < 0.001; n = 3. EMT: epithelial–mesenchymal transition; miR: microRNA; α-SMA: α-smooth muscle actin; RT-PCR: real-time polymerase chain reaction; SD: standard deviation.
COL10A1, COL12A1, and TGFβR1 are the targets of miR-101
TargetScan predicted COL10A1, COL12A1, and TGFβR1 as potential targets of miR-101 (http://www.targetscan.org). There were two miR-101 binding sites in the 3′-untranslated region (UTR) of human TGFβR1, and one in COL10A1 and COL12A1. We constructed luciferase reporter plasmid carrying a fragment of the COL10A1, COL12A1, or TGFβR1 3′-UTR that contained either the wild or mutated (Mut) type binding sites for miR-101. Results showed that miR-101 inhibited the luciferase activity of TGFβR1-T1 and TGFβR1-T2 but not the TGFβR1-T1-Mut and TGFβR1-T2-Mut, indicating that miR-101 only bound to wild type site in the 3′-UTR of TGFβR1. MiR-101 also suppressed the luciferase activity of wild type COL10A1 and COL12A1 3′-UTR (Figure 5(a)). MiR-101 had a higher inhibition on the luciferase activity of wild type COL10A1 than wild type COL12A1, so COL10A1 was used in further study. Further studies demonstrated that the transduction of HK-2 cells with a hsa-miR-101 mimic reduced the mRNA expression levels of COL10A1 and TGFβR1 (Figure 5(b)). Meanwhile, the forced expression of miR-101 inhibited the protein levels of COL10A1 and TGFβR1 in HK-2 cells (Figure 5(c)). These results suggested that COL10A1, COL12A1, and TGFβR1 were the targets of miR-101.

COL10A1, COL12A1, and TGFβR1 are the targets of miR-101. (a) Conserved miR-101-binding sites in the 3′-UTRs of COL10A1, COL12A1, and TGFβR1 mRNA are shown. Luciferase activity of the wild type or mutant COL10A1, COL12A1, and TGFβR1 is assessed in the 293T cells. (b) Effects of miR-101 mimics on the endogenous COL10A1 and TGFβR1 mRNA levels in HK-2 cells are determined by quantitative RT-PCR. (c) Effects of miR-101 mimics on the endogenous COL10A1 and TGFβR1 protein levels in HK-2 cells are determined by Western blotting. Data are presented as means ± SD, *p < 0.05, **p < 0.01, ***p < 0.001; n = 3. COL10A1, COL12A1: collagen, type X, alpha 1; TGFβR1: transforming growth factor β receptor 1; miR/mRNA: microRNA; RT-PCR: real-time polymerase chain reaction; SD: standard deviation.
Discussion
MiR-101 is often used as a tumor-inhibiting factor, and miR-101 expression level is decreased in many cancers because of genomic loss. A previous study has found that miR-101 suppresses proliferation and invasion of cancer cells by targeting enhancer of zeste homolog 2 (EZH2). 16 MiR-101 is an effective inhibitor of autophagy and enhances chemotherapeutic drug-induced apoptosis in cancer cells. 17 The different roles of miR-101 on pro-angiogenesis and anti-angiogenesis have been reported. 18,19 Besides, miR-101 promotes the production of lipopolysaccharide (LPS)-induced pro-inflammatory cytokine in macrophages via targeting mitogen-activated protein kinase phosphatase-1. 20 MiR-101 also suppresses liver and cardiac fibrosis. 9,21 In this study, we find that miR-101 level is downregulated in mice kidney tissues at 42 days after I/R injury. Impaired kidney function is associated with the progression of AKI-CKD as revealed by the increase of BUN and Scr levels at 35 days after I/R, which are in conformity to a previous study. 22 Our in vivo and in vitro evidence shows that AKI-CKD transition marker NGAL is highly expressed in I/R-induced renal tissues and hypoxic HK-2 cells with miR-101 downregulation. These data prove that miR-101 is involved in AKI-CKD transition. Furthermore, α-SMA expression is negatively regulated by miR-101 level. The loss of α-SMA attenuates tubulointerstitial fibrosis, indicating that miR-101 overexpression is renoprotective to mice.
In addition, EMT process of tubular epithelial cells plays an important role in renal fibrogenesis. During EMT, epithelial cells dedifferentiate and lose inherent features of epithelial cells, thereby inducing expression of mesenchymal markers and phenotypic alterations, resulting in the production of more fibroblast-like cells. 23 Tubular fibroblasts usually are induced to myofibroblasts during EMT due to the loss of E-cadherin expression and activation of α-SMA as well as vimentin. 24 Our in vivo studies show that miR-101 overexpression inhibits mRNA and protein expression levels of α-SMA and vimentin as well as increases E-cadherin expression in AKI-CKD transition, which is in line with a previous study. 11 Besides, Lovisa et al. have reported that in fibrotic renal parenchyma, renal tubules undergo atrophy or swelled, and tubular epithelial cells develop a series of molecular alterations like the abnormal expression of EMT markers. 25 Interstitial myofibroblasts are accumulated in kidney fibrosis and produce vast extracellular matrix, accompanied by collagen aggregation in the glomeruli and inflammatory cell infiltration. 26 Consistent with these findings, our renal histopathology displays collagen accumulation, renal interstitial fibrosis, fibroblast accumulation, and increased inflammatory cells during AKI-CKD transition. High expression of miR-101 greatly mitigates glomerular sclerosis, interstitial fibrosis, and inflammatory reaction. Our study suggests that miR-101 overexpression significantly protects renal tissues during EMT, which provides a reference for treatment of EMT-induced renal fibrosis.
TGFβR1 is a major EMT inducer by upregulating α-SMA expression and downregulating E-cadherin. 27 Huang et al. found that miR-101 inhibited TGFβR1-induced fibroblast activation by TGF-β/Smad2/3 signaling pathway, thereby inhibiting pulmonary fibrosis. 8 In the present study, miR-101 inhibits TGFβR1/COL10A1-induced fibroblast activation, characterized by the reduction of α-SMA and vimentin levels, as well as the increase of E-cadherin level. Therefore, COL10A1 is also considered to be a strong EMT inducer. Besides, COL10A1, COL12A1, and TGFβR1 are identified as the miR-101 targets by the luciferase reporter assays, and expression levels of COL10A1 and TGFβR1 are regulated by miR-101 levels. This is in agreement with a previous study in hepatic stellate cells. 21
Growing evidence indicates that hypoxia can induce differentiation of tubular cells into myofibroblasts, thereby accelerating renal fibrosis. Previous study indicates that Twist is related to the occurrence of EMT induced by hypoxia in renal tubular cells. 28 However, little is known about whether other molecules besides Twist play a role in hypoxia-induced EMT. Our reports show that miR-101 also regulates hypoxia-induced EMT in HK-2 cells, which is based on the following observations: (1) HK-2 cells exhibit myofibroblast-like characteristics under hypoxic conditions. HK-2 cells are observed the downregulation of E-cadherin expression and the upregulation of α-SMA and vimentin levels, which is in accordance with a previous report. 15 (2) The mRNA expression level of miR-101 is decreased in HK-2 cells after hypoxic exposure. However, miR-101 overexpression under hypoxic condition suppresses the hypoxic mesenchymal phenotype, exhibiting an increase in E-cadherin expression levels and a reduction in α-SMA and vimentin expression. This result further indicates that miR-101 may be an inhibitor of EMT in HK-2 cells. 29 (3) The expression level of miR-101 is positively correlated with E-cadherin. The mechanism of miR-101-mediated EMT may be because miR-101 promotes E-cadherin expression, thereby enhancing E-cadherin-mediated cell adhesion and inhibiting mesenchymal markers. 30
These evidences demonstrate that the upregulation or the downregulation of miR-101 levels is associated with kidney fibrogenesis and AKI-CKD progression, which could become a novel therapeutic means. Many studies find that lentivirus is widely used for the delivery of miRNAs in vivo to improve their activity. This therapeutic means has been validated in a mouse model of lung carcinoma 31 and pulmonary fibrosis. 8 In this study, lentivirus-mediated miR-101 kidney tissues alleviate the renal fibrosis in AKI-CKD progression, thereby improving renal function. Therefore, miR-101 may be a potential therapeutic miRNA for the treatment of renal fibrosis.
In summary, miR-101 possesses the ability to alleviate renal fibrosis after AKI. COL10A1, COL12A1, and TGFβR1 are the direct targets of miR-101. MiR-101 overexpression suppresses fibrosis and EMT process, thereby attenuating the progression of AKI-CKD. Although additional exploration is still needed, our study indicates that miR-101 is a novel therapeutic miRNA for the AKI-CKD progression.
Supplemental material
Supplemental Material, supplementary_Figure_1 - Upregulated miR-101 inhibits acute kidney injury–chronic kidney disease transition by regulating epithelial–mesenchymal transition
Supplemental Material, supplementary_Figure_1 for Upregulated miR-101 inhibits acute kidney injury–chronic kidney disease transition by regulating epithelial–mesenchymal transition by J-Y Zhao, X-L Wang, Y-C Yang, B Zhang and Y-B Wu in Human & Experimental Toxicology
Supplemental material
Supplemental Material, Title_and_description_for_supplemental_material - Upregulated miR-101 inhibits acute kidney injury–chronic kidney disease transition by regulating epithelial–mesenchymal transition
Supplemental Material, Title_and_description_for_supplemental_material for Upregulated miR-101 inhibits acute kidney injury–chronic kidney disease transition by regulating epithelial–mesenchymal transition by J-Y Zhao, X-L Wang, Y-C Yang, B Zhang and Y-B Wu in Human & Experimental Toxicology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Special Scientific Research Foundation for Traditional Chinese Medicine [Grant Number: 201507001-03].
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.