
Abstract
Chronic obstructive pulmonary disease (COPD), characterized by chronic inflammation, is a recognized global health crisis. Sialic acid-binding immunoglobulin-like lectin 1 (siglec1 or CD169), mainly expressed in macrophages and dendritic cells, is markedly upregulated after encountering pathogens or under acute/chronic inflammation conditions. However, it is rarely reported that whether siglec1 plays a role in the development of COPD. In this study, we found that siglec1 had higher expression in the lungs from COPD rats and in peripheral blood mononuclear cells (PBMCs) from COPD patients. Knockdown of siglec1 in vivo and in vitro dramatically decreased pro-inflammatory cytokines production in pulmonary macrophages and alleviated pulmonary inflammatory responses in COPD rats as well as inactivated nuclear factor kappa B (NF-κB) signaling. In addition, we identified a new microRNA, miR-195-5p, which has never explored in COPD, was lower expressed in COPD rats and PBMC of COPD patients, and could negatively modulate siglec1 expression in macrophages. Moreover, overexpression of miR-195-5p via miR-195-5p mimics in vitro and in vivo could significantly alleviate pro-inflammatory cytokines production in pulmonary macrophages and pulmonary inflammatory responses in COPD rats. Together, our findings suggested that miR-195-5p inhibited the development of COPD via targeting siglec1, which might become a therapeutic target to improve COPD.
Keywords
Introduction
Chronic obstructive pulmonary disease (COPD) is one of the biggest emerging crisis to global health. Alarmingly, it is estimated that nearly 80 million individuals suffered with moderate-to-severe COPD. Generally, chronic inflammation and obstructed airflow are the common clinical features of COPD. 1 Many studies have shown that various factors contribute to the development and pathogenesis of COPD, such as genetic factors, environmental factors, host immune status, and the balance between pro-ant and antioxidant system. 2,3 Of note, cigarette smoke (CS) is generally regarded as an important contributor for the development of COPD. However, the nature of the pathogenesis of COPD and the underlying mechanisms linking COPD with its comorbidities are still not completely clear.
The sialic acid-binding immunoglobulin-like lectin 1 (siglec1 or CD169) is an interferon-inducible cell surface receptor, mainly expressed in macrophages and dendritic cells. 4,5 siglec1 is upregulated after encountering pathogens or under acute/chronic inflammation conditions. 6 Previous studies have demonstrated that siglec1 is involved in inflammatory immune responses after pathogens infection, such as retroviruses and Campylobacter jejuni. 7 Simultaneously, siglec1 is recently reported to associate with development of asthma and susceptibility to pulmonary active tuberculosis. 8,9 Moreover, siglec1 is highly associated with inflammation-related diseases, such as type 1 diabetes and systemic lupus erythematosus. 10,11 However, whether siglec1 participates in the development of COPD is rarely reported.
MicroRNAs (miRNAs) are short, non-coding RNAs that can regulate targeted gene expression via binding the 3′ untranslated regions (3′UTRs). Many studies have reported that miRNAs perform critical roles in diverse pathological processes such as cell proliferation, apoptosis, and metastasis. 12,13 Aberrant expressions of miRNAs are associated with many diseases such as cancers, infection, and autoimmune diseases. 14,15 It has been demonstrated that miRNAs are involved in the modulation of inflammatory immune responses, which is a characteristic in many lung diseases such as asthma and COPD. 16 Recently, many miRNAs have been identified to be associated with the pathogenesis of COPD such as miR-31, miRNA-145, miR-29b, and miRNA-338. 17 –19 In the present study, we identified miR-195-5p, which was lower expressed in COPD rats and PBMC of COPD patients. Additionally, miR-195-5p controlled host inflammatory immune responses via negatively modulating siglec1 expression in macrophages. Data indicate that miR-195-5p inhibited the development of COPD via targeting siglec1, suggesting that miR-195-5p might become a therapeutic target to improve COPD.
Materials and methods
Animals
Forty-eight male Wistar rats (8-week-old) used in this study were purchased from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). All rats were housed under specific pathogen-free conditions at Luohe Central Hospital (Henan, China). All animal experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Luohe Central Hospital (approval no. IEC-2017060601).
PBMC isolation
Peripheral blood mononuclear cells (PBMCs) from healthy volunteers and COPD patients (Department of Pharmacy, Luohe Central Hospital, China) were isolated as previously reported. 20 In brief, peripheral blood was diluted with an equal volume of phosphate-buffered saline (PBS), pH 7.4. Then, 15 ml of diluted blood was layered over 30 ml of the Ficoll (GE Healthcare, Shanghai, China) based on the manufacturer’s protocol and centrifuged at 400 × g for 30 min at room temperature. The PBMC interface was removed and washed with PBS. PBMC pellets were suspended RMPI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) for the following experiments. All experiments in this study were conducted based on the protocols approved by the Department of Pharmacy of Luohe Central Hospital (Henan, China).
Establishing a rat model of COPD
We established a rat model of COPD by combining CS with lipopolysaccharide (LPS). 21 After acclimation for 3 days, rats were randomly divided into two groups (n = 12 in each group): (1) COPD model group: on day 1, day 15, and day 28, each rat received intratracheal instillation of LPS (200 μg/kg) after anesthesia with 5% isoflurane. While from days 2 to 27 (except day 15), the rats were exposed to CS from eight continuously burned cigarettes for 30 min in a covered box (70 × 60 × 60 cm3). (2) Control group: rats did not receive LPS or smoke exposure for 30 days. Commercial cigarettes were provided by Shanghai Tobacco Industry Co., Ltd (Shanghai, China). Finally, rats were subjected to a laparotomy during anesthesia and euthanized via exsanguination through section of the abdominal aorta. The trachea was clamped at the end of inspiration and the lungs were collected for further analysis.
For siglec1 knockdown or miR-195-5p overexpression in vivo, we injected adenovirus containing shRNA targeting siglec1 (Ad-shRNA) or miR195-5p mimics (Ad- miR-195-5p mimics) (Hanbio Biotechnology Co. Ltd, Shanghai, China) with 1 × 108 viral particles in COPD rats. Thus, another 24 rats were randomly divided into 6 different groups (n = 4) in our study, including Ad-sh-NC + Normal lungs, Ad-sh-Siglec1 + Normal lungs, Ad-sh-NC + COPD lungs, Ad-sh-Siglec1 + COPD lungs, Ad-miR-NC and Ad-miR-195-5p mimic.
Cell culture
Pulmonary macrophage cell lines (NR8383) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, California, USA) supplemented with 10% FBS (Gibco, MD, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in humidified air atmosphere with 5% CO2.
Western blot
Pulmonary macrophages (NR8383) and lung tissues from COPD rats were lysed with radio immunoprecipitation assay lysis buffer with protease inhibitors. Protein concentration was measured using bicinchoninic acid protein assay kit. The equal amount of protein in each sample was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, USA) and then transferred to polyvinylidene difluoride membranes (Bio-Rad). Blocked by 5% nonfat dry milk in Tris-buffered saline with Tween® 20 (TBS-T; Sigma-Aldrich, St. Louis, MO, USA), and the membranes were incubated with primary antibodies overnight at 4°C. Subsequently, membranes were washed with TBS-T and then cultured with specific secondary antibodies for 1 h. Primary antibodies against siglec1 (ab53443, Abcam, Cambridge, MA, USA), tumor necrosis factor alpha (TNF-α; ab1793, Abcam), interleukin 1 beta (IL-1β; ab9722, Abcam), IL-6 (ab6672, Abcam), IL-8 (ab18672, Abcam), p65 (ab16502, Abcam), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; ab245356, Abcam) were employed separately. Signals were developed by Pierce enhanced chemiluminescence Western blotting substrate (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Hematoxylin and eosin staining
Lung tissues were fixed with 4% paraformaldehyde for 48 h and embedded in paraffin according to standard procedures. 22 Then, the lung tissues were sectioned at 4 µm thickness and deparaffinized with xylene. Finally, the sections were stained with hematoxylin and eosin (HE) and scanned using Nikon microscope for histological analysis.
Real-time PCR
Total RNA from pulmonary macrophages (NR9393) and PBMCs were isolated using TRIzol (Invitrogen) based on the previous study. 23 Then, 1 µg RNA was reversely transcribed using PrimeScript™ RT Master Mix (Takara, Dalian, China, RR036A). Real-time polymerase chain reaction (PCR) was performed using Power SYBR Green PCR Master Mix (Takara, RR420A). In this study, we used GAPDH or U6 as respective internal controls to normalize the target genes transcript levels based on 2−ΔΔC t method. The primers used in this study are listed in Table 1.
Primers used in this study.

TNF-α: tumor necrosis factor alpha, IL-1β: interleukin 1 beta; Siglec1: sialic acid-binding immunoglobulin-like lectin 1; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; miR-195-5p: microRNA.
Cytokines measurement
Pulmonary macrophages (NR8383) culture supernatants and rat serum isolated from 0.1 ml blood sample were collected and stored at −20°C until analysis. The concentration of TNF-α, IL-6, IL-1β, IL-8, and NF-κB (p65) levels were measured by SET-Ready-GO enzyme-linked immunosorbent assays (ELISA) kits (eBioscience, San Diego, CA, USA) according to the manufacturer’s protocol.
Luciferase reporter assays
Luciferase assays were performed as described previously. 24 In brief, pulmonary macrophages (NR8383) were transfected with reporter plasmids derived from pGL3 containing 3′UTR inserted 5′-upstream of firefly luciferase and a control reporter plasmid. Simultaneously, pulmonary macrophages were transfected with miR-195-5p mimics and inhibitors. The transfected cells were cultured for 24 h, and then were collected and lysed. Firefly and Renilla luciferase activity were detected via using Dual-Luciferase Reporter System (Promega, Madison, Wisconsin, USA). Finally, Renilla luciferase activity was utilized as a control to normalize for transfection efficiency.
Statistical analysis
All statistical analyses were performed using SPSS17.0 software. The data were subjected to one-way analysis of variance, followed by the Tukey’s test. A value of p < 0.05 was considered to be statistically significant. The results were expressed as mean ± standard error of the mean based on three indicated experiments.
Results
Higher pulmonary siglec1 expression in PBMC from COPD subjects
To determine the role of siglec1 in the pathogenesis of COPD, we firstly analyzed the expression of siglec1 in PBMC from normal or COPD subjects via bioinformatics analysis (GSE 52509-GEO2 R). From the results, we observed that compared with normal subjects, the expression of siglec1 was markedly upregulated in COPD lungs of mouse (Figure 1(a)). In addition, we also determined the expression of siglec1 in human PBMC from normal volunteers or smokers, showing that siglec1 expression was higher in PBMC from smokers than from normal volunteers (Figure 1(b)). To further confirm siglec1 expression in PBMC from COPD subjects, we generated COPD model in rats by combining CS with LPS. HE staining showed the alveolar structure was intact in the normal group, whereas in COPD group, there were emphysematous changes, the alveolar size was different, the alveolar number was decreased, the alveolar space was enlarged, and the alveolar septum was fractured (Figure 1(c)), suggesting that COPD model was successfully generated. Moreover, the mean linear intercept (MLI) in the COPD group was significantly larger than the control group (Figure 1(d)). Next, we detected the expression of siglec1 in the lung of COPD rats and confirmed higher siglec1 protein level in lung from COPD rats (Figure 1(e)). Together, siglec1 expression is significantly increased in COPD subjects.
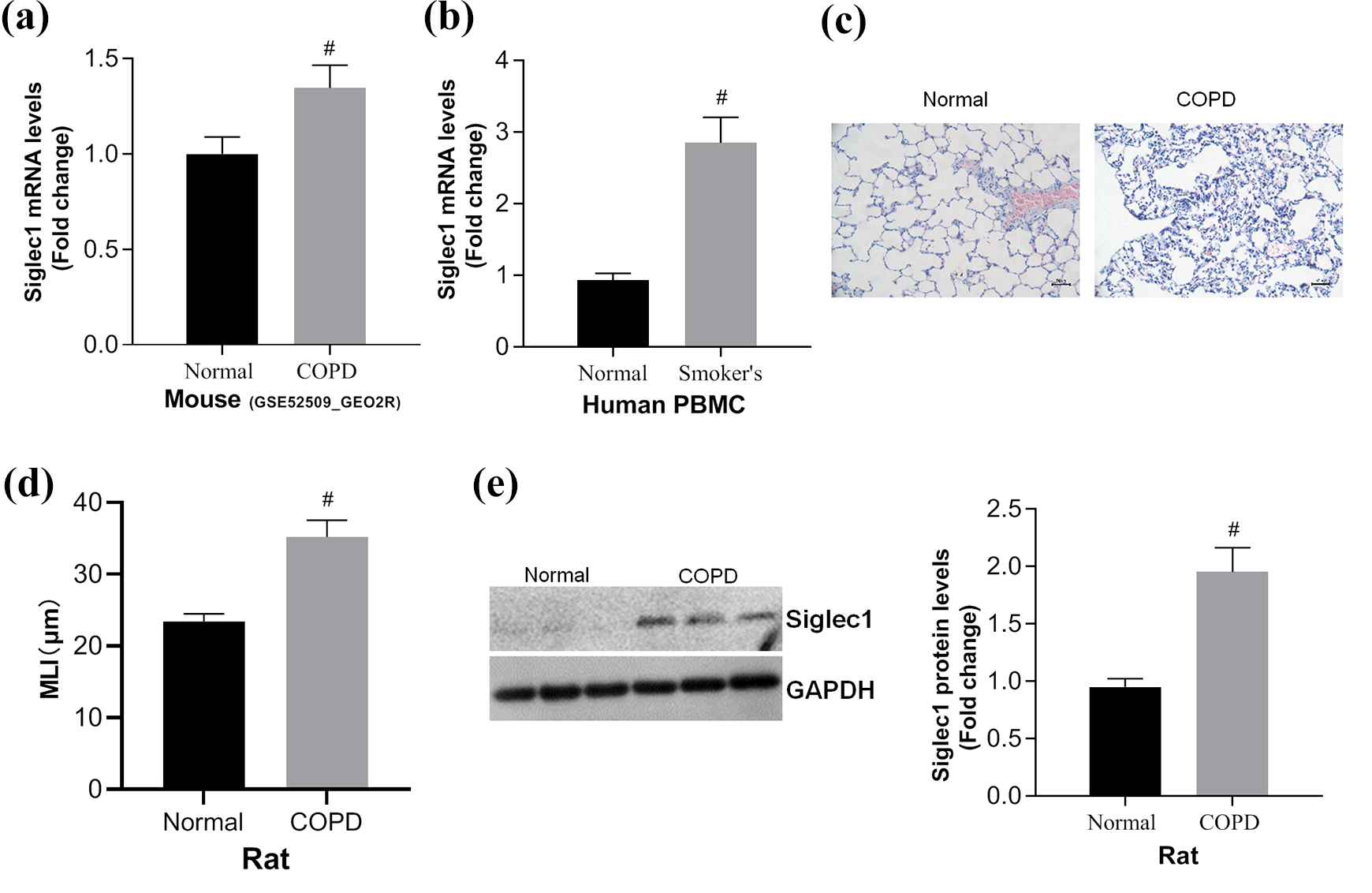
Higher pulmonary siglec1 expression in COPD. (a) Bioinformatics analysis showed higher pulmonary siglec1 expression in mouse COPD models. (b) RT-qPCR revealed the expression of siglec1 in PBMC from normal volunteers and COPD patients. (c) HE staining of lung from control and COPD rats. (d) The MLI was evaluated in control and COPD groups. (e) Western blot analysis detected the expression of siglec1 in lung from COPD rats. #p < 0.05: compared with NC or normal group. siglec1: sialic acid-binding immunoglobulin-like lectin 1; COPD: chronic obstructive pulmonary disease; RT-qPCR: real-time quantitative polymerase chain reaction; PBMC: peripheral blood mononuclear cell; HE: hematoxylin and eosin; MLI: mean linear intercept; NC: normal control.
Knockdown siglec1 alleviates inflammatory immune responses in COPD subjects
Many studies have demonstrated that inflammatory immune responses play crucial roles in modulation of COPD progression. Thus, we also determined whether siglec1 could regulate host inflammatory responses in COPD. We used adenovirus containing shRNA targeting siglec1 (Ad-sh-siglec1c) in COPD rats to knockdown siglec1 in vivo. As shown in Figure 2, siglec1 silence had no significant influence on the concentration and protein levels of pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and IL-8 in normal lungs. However, compared with COPD rats, knocking down of siglec1 significantly decreased the concentration of TNF-α, IL-1β, IL-6, and IL-8 (Figure 2(a) to (d)). The activation of NF-κB signaling is widely proposed to affect the development of diseases. Simultaneously, we also determined the effects of knockdown siglec1 on NF-κB signaling and found that knockdown of siglec1 significantly decreased NF-κB (p65) activation compared with COPD rats (Figure 2(e)). In addition, the protein levels of TNF-α, IL-1β, IL-6, IL-8, and p65 were evaluated by Western blot analysis, showing that the protein levels of TNF-α, IL-1β, IL-6, IL-8, and p65 were increased in COPD rats, but deficiency of siglec1 counteracted the results (Figure 2(f) and (g)). In summary, knockdown siglec1 alleviates inflammatory immune responses in COPD subjects.

Knockdown of siglec1 inhibited inflammatory immune responses in COPD rats. COPD models were established via combining cigarette smoke with LPS in rats. Siglec1 was knockdown via injecting 1 × 108 Ad-sh-Siglec1 viral particles in COPD rats, and after 15 days, lungs were sampled for parameters analysis. (a to e) ELISA showed the concentration of TNF-α, IL-1β, IL-6, IL-8, and NF-κB in lung from COPD rats with/without injection of Ad-sh-Siglec1 viral particles. (f and g) Western blot analysis assessed the protein expression of TNF-α, IL-1β, IL-6, IL-8, and NF-κB in lung from COPD rats with/without injection of Ad-sh-Siglec1 viral particles. #p < 0.05: compared with NC or normal lung. siglec1: sialic acid-binding immunoglobulin-like lectin 1; LPS: lipopolysaccharide; ELISA: enzyme-linked immunosorbent assay; TNF-α: tumor necrosis factor alpha, IL-1β: interleukin 1 beta; NF-κB: nuclear factor kappa B.
miR-195-5p negatively regulates siglec1 expression in COPD subjects
Next, we explored why siglec1 was upregulated in COPD subjects. Many studies have shown that miRNAs may perform roles in regulating siglec1 expression. 25 Thus, we firstly used bioinformatics analysis based on Targetscan and miRBase to predict some of miRNAs (Figure 3(a)) that may regulate siglec1 expression. Thereafter, we synthesized mimics of these miRNAs and transfected these miRNAs into pulmonary macrophages (NR8383 cells). As shown in Figure 3(a), almost all miRNAs mimics could downregulate siglec1 expression, but the efficiency of miR-195-5p was the most significant, suggesting that miR-195-5p may be critical for regulating siglec1 expression in COPD. Subsequently, we compared the expression of miR-195-5p in PBMC between the normal volunteers and the smokers using bioinformatics analysis and found that the expression of miR-195-5p was lower in smoker’s lungs (Figure 3(b)). Further, we detected the mRNA abundance of miR-195-5p in PBMC from normal and smoker volunteers, and confirmed lower expression of miR-195-5p in smoker’s lungs (Figure 3(c)). In addition, we determined the expression of miR-195-5p in lung from normal or COPD rats and further verified the low expression of miR-195-5p in COPD subjects (Figure 3(d)). To determine whether miR-195-5p could regulate siglec1 expression, we transfected NR8383 cells with miR-195-5p mimic and inhibitor. As shown in Figure 3(e), transfection with miR-195-5p mimic upregulated miR-195-5p expression; in contrast, transfection with miR-195-5p inhibitor significantly decreased miR-195-5p expression. Simultaneously, we generated luciferase reporter system targeted 3′UTR of siglec1 and found that transfection with miR-195-5p mimic markedly decreased the luciferase activity of siglec1-WT while miR-195-5p inhibitor exhibited opposed effects. However, in the deletion or mutation group of siglec1, the luciferase activity had no evident changes in all groups (Figure 3(f)). Next, by using Western blot, we confirmed miR-195-5p could negatively regulate the siglec1 protein expression in NR8383 cells (Figure 3(g) and (h)). To sum up, miR-195-5p negatively regulates siglec1 expression in COPD subjects.
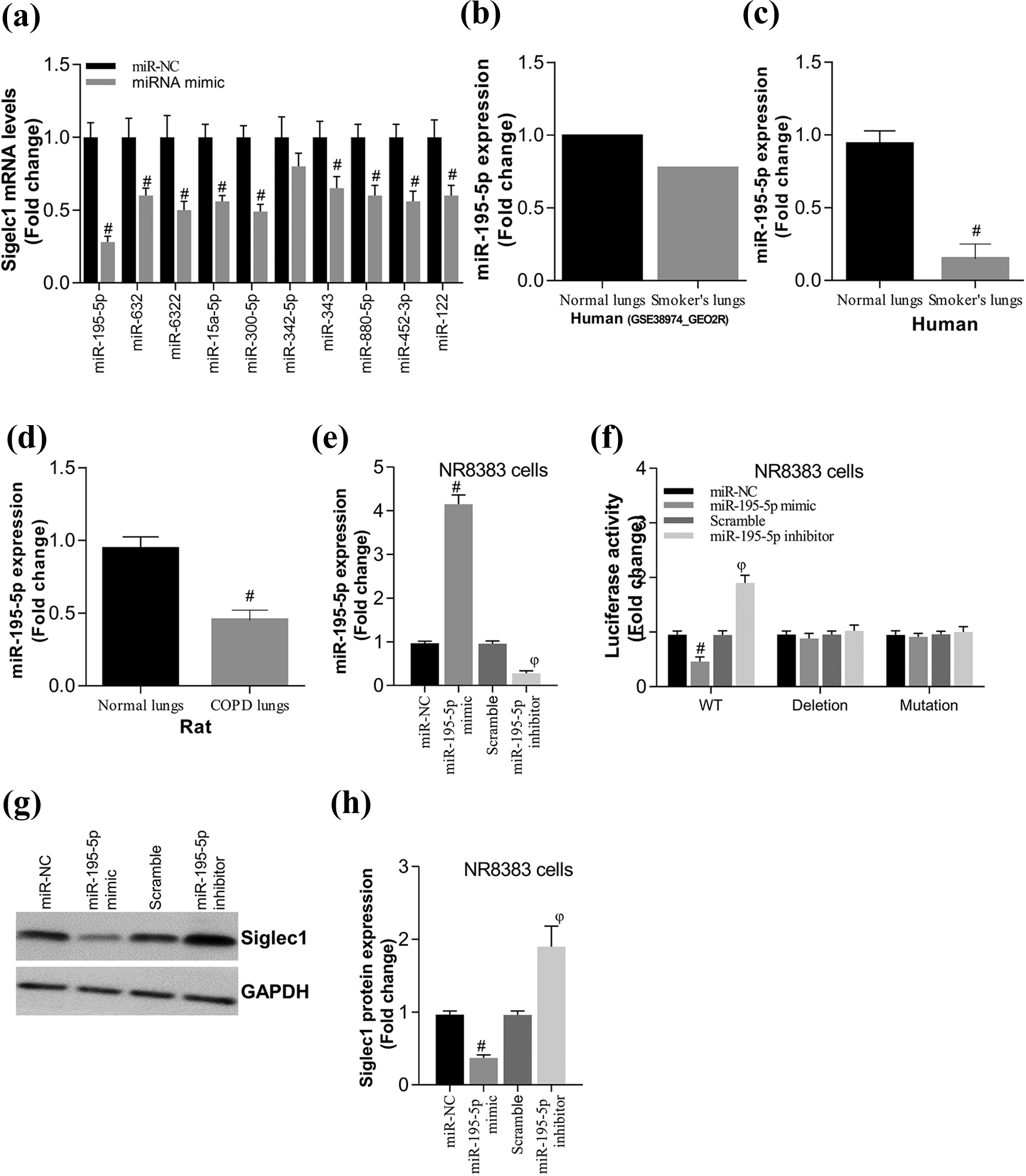
MiR-195-5p negatively regulated siglec1 expression in pulmonary macrophages. (a) Bioinformatics analysis based on Targetscan and miRBase to predict miRNAs targeted siglec1, and RT-qPCR assessed siglec1 expression in NR8383 cells after overexpression of miRNA mimics. (b) The expression of miR-195-5p in PBMC from COPD patients via bioinformatics analysis. (c and d) The mRNA abundance of miR-195-5p in PBMC from COPD patients (c) and COPD rats (d) via qPCR. (e) RT-qPCR was applied to estimate the expression of miR-195-5p in NR8383 cells after transfection with miR-195-5p mimic and inhibitor. (f) Luciferase reporter assays determined the effects of transfection with miR-195-5p mimic and inhibitor in stability of 3′UTR of siglec1 in NR8383 cells. (g and h) Siglec1 protein level was examined in NR8383 cells after transfection with miR-195-5p mimic and inhibitor using Western blot analysis. #p < 0.05: compared with miR-NC or normal group; ϕp < 0.05: compared with scramble group. miRNAs: MicroRNA; RT-qPCR: real-time quantitative polymerase chain reaction; PBMC: peripheral blood mononuclear cell; COPD: chronic obstructive pulmonary disease; 3′UTR: 3′ untranslated regions; NC: normal control.
Overexpression of miR-195-5p inhibits inflammatory responses in lung of COPD rats and NR8383 cells
To determine the physiological function of miR-195-5p in COPD, we generated ad-miR-195-5p viral particles and injected it via vein to overexpress miR-195-5p in vivo in COPD rats. We found that overexpression of miR-195-5p could alleviate damage to lungs of COPD rats (Figure 4(a)). In addition, the MLI was overtly lower by miR-195-5p overexpression (Figure 4(b)). Simultaneously, the mRNA and concentration as well as the protein expression of pro-inflammatory genes including TNF-α, IL-1β, IL-6, and IL-8 were significantly decreased in the lungs from COPD rats by upregulation of miR-195-5p (Figure 4(c) to (i)). Expectedly, overexpression of miR-195-5p decreased p65 mRNA, and concentration and protein levels, too (Figure 4(c) and (i)), indicating that miR-195-5p suppressed NF-κB activation in COPD subjects compared with the control groups. Next, we transfected pulmonary macrophages (NR8383 cells) with miR-195-5p mimic, after smoke extract stimulation, we found that overexpression of miR-195-5p significantly decreased the concentration and protein levels of TNF-α, IL-1β, and p65 (Figure 4(j) to (n)). On the whole, overexpression of miR-195-5p inhibits inflammatory responses in the lungs of COPD rats and NR8383 cells.

Overexpression of miR-195-5p inhibited inflammatory responses in lung from COPD rats. (a) HE staining of lung from COPD rats with/without overexpression of miR-195-5p via injecting Ad-miR-195-5p viral particles. (b) MLI was assessed in different groups. (c) RT-qPCR uncovered the mRNA abundance of pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, IL-8, and NF-κB in lung from COPD rats with/without injection of Ad-miR-195-5p viral particles. (d to h) ELISA determined pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, IL-8, and NF-κB in serum from COPD rats with/without injection of Ad-miR-195-5p viral particles. (i) Western blot detected TNF-α, IL-1β, and p65 levels in lung from COPD rats with/without injection of Ad-miR-195-5p viral particles. (j to l) ELISA determined the concentration of TNF-α, IL-1β, and p65 in NR8383 cells with/without overexpression of miR-195-5p after the smoke extract stimulation. (m and n) Western blot analysis detected TNF-α, IL-1β, and NF-κB levels in NR8383 cells with/without overexpression of miR-195-5p after smoke extract stimulation. ϕp < 0.05: compared with miR-NC group. HE: hematoxylin and eosin; miR-195-5p: microRNA; MLI: mean linear intercept; RT-qPCR: real-time quantitative polymerase chain reaction; TNF-α: tumor necrosis factor alpha, IL-1β: interleukin 1 beta; NF-κB: nuclear factor kappa B; ELISA: enzyme-linked immunosorbent assay.
MiR-195-5p attenuates pro-inflammatory responses in COPD via targeting siglec1
To explore whether miR-195-5p attenuated inflammatory responses through negatively regulating siglec1 expression, we overexpressed siglec1 in NR8383 cells transfected with miR-195-5p mimic after smoke extract stimulation. Consistently, miR-195-5p mimic significantly decreased TNF-α, IL-1β, and p65 concentration and protein levels (Figure 5(a) to (c)). However, overexpression of siglec1 reversed these effects induced by miR-195-5p mimic, manifesting that siglec1 significantly increased TNF-α, IL-1β, and p65 concentration and protein levels (Figure 5(a) to (c)). Taken together, miR-195-5p attenuates pro-inflammatory responses in COPD via targeting siglec1.

miR-195-5p inhibited inflammatory immune responses in COPD via targeting siglec1. (a) ELISA determined TNF-α, IL-1β, and NF-κB concentration in NR8383 cells transfected with miR-195-5p mimics together with/without siglec1 vectors after the smoke extract stimulation. (b and c) Western blot analysis detected TNF-α, IL-1β, and NF-κB expression in NR8383 cells transfected with miR-195-5p mimics together with/without siglec1 vector after smoke extract stimulation. #p < 0.05: compared with miR-NC + pcDNA group; ϕp < 0.05: compared with miR-195-5p mimic + pcDNA group. miR-195-5p: microRNA; COPD: chronic obstructive pulmonary disease; ELISA: enzyme-linked immunosorbent assay; TNF-α: tumor necrosis factor alpha, IL-1β: interleukin 1 beta; NF-κB: nuclear factor kappa B.
Discussion
COPD is a multifactorial disease which is a great threat to public health. Many studies have demonstrated that environmental and genetic risk factors can contribute to the pathogenesis of COPD. 26 In this study, we found that siglec1 was higher expressed in PBMC from COPD rats and patients, and siglec1 promoted the development of COPD via exacerbating pulmonary inflammatory immune responses in macrophages. Additionally, we identified a new miRNA, miR-195-5p, which was lower expressed in COPD rats and patients and could alleviate the development of COPD via negatively modulating siglec1 expression. To the best of our knowledge, our study provides the first evidence for linking siglec1 and miR-195-5p in the development of COPD.
Generally, chronic inflammation is a dominant feature of COPD. In these processes, a number of inflammatory cell types including macrophages and neutrophils have contributed to the pathogenesis of COPD. 27 These cells release various inflammatory cytokines such as TNF-α, IL-6, IL-1β, and IL-8 and result in impairment in clearance of apoptotic cells and, therefore, potentially contribute to the chronic inflammatory state in the lungs in response to CS, 28 –30 suggesting that macrophages perform critical roles in the development of COPD. 31,32 Siglec1 is a receptor sialic acid-tagged pathogen or other antigen and is mainly expressed in monocytes/macrophage. Upon recognition of glycan ligands, siglec1 is activated and then modulates the host immune balance in infection, sepsis, autoimmune diseases, and cancers. 6 Previous studies have demonstrated that CD169+ macrophages, mainly localized distantly from epithelial border, can exacerbate inflammation by producing CCL8 that recruits inflammatory monocytes after mucosal injury, suggesting that siglec1 may facilitate inflammation-mediated damages via changing macrophage’s function. 4,33 Indeed, we found siglec1 was highly expressed in COPD rats and patients. Moreover, knockdown of siglec1 alleviated pulmonary inflammatory immune responses and decreased NF-κB activation in COPD rats. These data suggested that siglec1 could promote COPD development via increasing pro-inflammatory cytokines production and activating NF-κB signaling in macrophages. As far as we know, our study is the first report to demonstrate siglec1 is associated with the pathogenesis of COPD.
Next, we explored how and why siglec1 was upregulated in COPD patients and rats. Many ongoing studies have demonstrated that dysregulated miRNAs are associated with COPD pathogenesis, and even miRNAs have the potential of being used as biomarkers for COPD. 34 Meantime, the studies have shown that miRNAs may perform roles in regulating siglec1 expression. Thus, we speculated that miRNAs may regulate siglec1 expression to regulate COPD progression. 25 To verify this hypothesis, we screened miRNAs that may target siglec1 via bioinformatics analysis. Then, we transfected these miRNA mimics into cells and found that miR-195-5p was the most efficient to downregulate siglec1 expression in macrophages. In addition, we generated luciferase reporter system and further confirmed miR-195-5p could regulate siglec1 expression by targeting 3′UTR of siglec1. However, siglec1 is an interferon-inducible receptor and can be upregulated on monocytes and macrophage upon interferon alpha (IFN-α) stimulation. Simultaneously, the expression of siglec1 is upregulated in different IFN type I-driven diseases, such as type 1 diabetes. 35 Thus, we do not exclude the role of interferon in regulating siglec1 expression in COPD, because IFN-α/β is associated with the development of some lung diseases such as infection and asthma. 36 Moreover, we overexpressed miR-195-5p via injection of Ad-miR-195-5p viral particles in vein in COPD rats and found that overexpressed miR-195-5p significantly alleviated pulmonary inflammation and decreased pulmonary damages, suggesting that miR-195-5p may be a potential therapeutic target to improve COPD.
In conclusion, we found that siglec1 could promote the development of COPD via regulating pro-inflammatory cytokines production in macrophages in lungs. Moreover, we identified miR-195-5p could negatively modulate siglec1 expression in macrophages. Simultaneously, overexpression of miR-195-5p could alleviate pulmonary inflammatory immune responses and damages in COPD rats. Thus, our data firstly demonstrated that miR-195-5p regulate siglec1 expression and finally control inflammation in lungs of COPD. Together, these findings indicate that miR-195-5p has the potential to improve the standard therapeutic approaches to COPD.
MiR-195-5p was previously confirmed to suppress the progression of melanoma through targeting hTERT. 37 MiR-195-5p blocks the cervical carcinoma cell migration and invasion via targeting ARL2. 38 MiR-195-5p alleviates the development of non-small-cell lung cancer by inhibiting the expression of CEP55. 39 However, the role and function of miR-195-5p in COPD was obscure.
Footnotes
Acknowledgments
We appreciate the support of Institutes from Luohe Central Hospital, The First Affiliated Hospital with Nanjing Medical University, Huadong Research Institute for Medicine and Biotechnics, and School of Life Science and Technology, China Pharmaceutical University.
Author contributions
SL and LJ contributed equally to this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Youth Top Talent Foundation of Luohe (2018QNBJRC01005) and National Natural Science Foundation of China (81573213).