
Abstract
This study investigated the effect of carvedilol on aspirin-induced gastric damage. Male Wistar rats were divided into three groups. Control rats received the vehicle, while the aspirin group received aspirin (200 mg/kg) orally for 4 days. Rats of aspirin + carvedilol group were administered aspirin along with carvedilol (5 mg/kg; intraperitoneal) for 4 days. Animals were euthanized at the end of the treatment period, and gastric tissues were collected to perform histopathological and mechanistic studies. The results revealed that aspirin administration induced gastric ulcer as there were remarkable histopathological lesions in the form of marked necrosis, inflammation, hemorrhage, edema, and dysplastic changes. Lipid peroxidative markers such as malondialdehyde, 4-hydroxynonenal, and protein carbonyl were significantly elevated in the aspirin group. This was concurrent with a significant amelioration of antioxidants such as reduced glutathione, superoxide dismutase, and catalase. Furthermore, aspirin increased the immunoexpression of cyclooxygenase (COX) 2 and nuclear factor kappa-B (NF-κB). Aspirin induced elevation in the inflammatory cytokines such as tumor necrosis factor-α, interleukin-6, and interleukin-1β. Aspirin enhanced the immunoexpression of inducible nitric oxide synthetase (iNOS) and increased the level of nitrite/nitrate in gastric tissue. On the other hand, carvedilol treatment reversed all these pathological changes. Carvedilol succeeded to enhance antioxidants in gastric tissue, attenuated lipid peroxidative parameters, and suppressed the release of inflammatory mediators. It attenuated the immunoexpression of COX-2, NF-κB, and iNOS. Collectively, carvedilol has a gastro-protective effect that could be attributed to its antioxidative and anti-inflammatory properties, which modulate NF-κB/COX-2/iNOS pathways.
Introduction
Gastric ulcer is a common gastrointestinal tract (GIT) disorder that affects about 10% of the world population. This pathological condition is characterized by GIT bleeding, perforation, and erosion of the mucosa wall due to imbalance between aggressive (acid, pepsin, and Helicobacter pylori) and defensive factors (mucin, prostaglandin (PG), bicarbonate, nitric oxide (NO), mucosal blood flow, and growth factors). 1,2 The incidence of this disease is multifactorial, which includes H. pylori, nonsteroidal anti-inflammatory drugs (NSAIDs), smoking, stress, chronic alcohol, altered PG E series metabolism, and bad dietary habits. 3
Aspirin is acetylsalicylic acid that has been used for last several decades as analgesic, antipyretic, and anti-inflammatory agent against multiple types of inflammation and in the prevention of cardiovascular thrombotic diseases. 4 Despite its therapeutic benefits, the use of aspirin is a major problem due to association with gastric ulcer. 5 The pathogenesis of aspirin-induced gastric ulceration includes the aspirin inhibiting the activities of the cyclooxygenase (COX) leading to decrease in PGs, which may be associated with reduced mucus and bicarbonate secretion, decreased mucosal blood flow, impaired platelet aggregation, alteration of microvascular structures leading to epithelia damage, increased leukocyte adherence and increased production of reactive oxygen species (ROS), increased lipid peroxidation and neutrophil infiltration, and decreased antioxidant enzymes. PG-independent pathways involving trapping of drug in epithelial cells, uncoupling of mitochondrial phosphorylation, lipid peroxidation, and superoxide generation also contribute to pathogenesis of gastric injury. 6 Furthermore, it has been reported that increase in NO synthase (NOS) activity is involved in the gastrointestinal mucosal defense and in the pathogenesis of mucosal damage. 7,8
Carvedilol is an antihypertensive agent that is commonly used in the treatment of arterial hypertension, heart failure, and angina pectoris based on its combined β- and α1-blocking activities. However, its therapeutic effect is associated with its antioxidant and anti-peroxidative properties. It was shown that carvedilol acts as a metal scavenger and can protect mitochondria against the oxidative damage. 9 –11 Furthermore, recent studies have elucidated the protective anti-oxidative and anti-inflammatory activities of carvedilol against nephro-, hepato-, and cardiotoxicity. 12 –15 However, its protective effects on aspirin-induced gastric ulcer or gastrointestinal toxicity has not been tested yet. Therefore, this study was designed to investigate the gastro-protective effects of carvedilol against aspirin-induced gastric ulcers in rats and to elucidate its possible mechanisms.
Materials and Methods
Animals
Male Wistar rats at 8 weeks of age (weighing 150–200 g) were obtained from the animal facility of College of Pharmacy, Taibah University. Prior to the commencement of the study, animals were acclimated in an air-conditioned room at 25°C, 12 h light/dark cycle, and 50 ± 55% humidity. Animals were allowed standard chow and free access to drinking water. The experimental protocol was approved by Research Ethics Committee of Taibah University (Reference number: TUCD-REC/20130313), which adopts and adheres to the guidelines of National Institutes of Health (NIH).
Chemical and reagents
Aspirin, carvedilol, and carboxymethylcellulose sodium salt (CMC) were purchased from Sigma Chemical (St. Louis, Missouri, USA).
Experimental design
A total of 24 rats were divided into three groups (8 animals in each group) each as follows: Group I: Animals in this group received 3 mL of 1% CMC by gavage. Group II: Animals received aspirin (200 mg/kg) orally suspended in 3 mL of 1% CMC for 4 days. Group III: Animals were administered aspirin (200 mg/kg, orally) along with carvedilol (5 mg/kg, intraperitoneal).
At the end of day 4, the animals were anesthetized by ketamine and the blood samples were collected from the heart. Animals were then euthanized, and stomach sections were isolated for determining the histopathological lesions and the analysis of oxidative and inflammatory parameters. 16
Estimation of histological lesions
Gastric tissue samples from each group were fixed in 10% neutral-buffered formalin for 24 h. The specimens were then embedded in paraffin, sectioned, and stained with hematoxylin and eosin, before being evaluated by light microscopy. Scoring for gastric damage, from 0 to 4, was made as previously described. 17 In brief, 0 refers to no lesions; 1 means changes limited to disruption of the surface lining epithelium or superficial layer of the mucosa, with no vascular congestion; 2 half of the mucosal thickness showing tissue necrosis; 3 more than two-thirds of the mucosal thickness destroyed, with marked tissue necrosis and vascular congestion, the muscular mucosa remained intact; and 4 complete destruction of the mucosa with necrosis and hemorrhage.
Estimation of lipid peroxidation and antioxidant system
Malondialdehyde (MDA) content
The MDA concentration, as an indicator of lipid peroxidation in the stomach tissue, was measured in the supernatants of the gastric homogenate using a commercially available kit (Bio Diagnostic Co., Giza, Egypt), according to the method described. 18 The amount of MDA formed was quantitated by reaction with thiobarbituric acid (TBA) and used as an index of lipid peroxidation. Absorption of the pink supernatant was measured spectrophotometrically at 532 nm. The amount of TBA reactive substances was expressed as nanomole MDA per gram wet tissue.
4-Hydroxynonenal (4-HNE) and protein carbonyl (PC) contents
They were estimated in the supernatants of the gastric homogenates according to the instructions of enzyme-linked immunosorbent assay (ELISA) kits (MyBioSource, San Diego, California, USA).
Reduced glutathione (GSH) content
GSH content was determined in liver homogenate supernatant using a commercial kit purchased from Bio Diagnostic Company. 19 Briefly, GSH reacts with 5, 5-dithiobis-2-nitrobenzoic acid, and the product has a maximal absorbance at 412 nm at spectrophotometer. The results were expressed as nanomole GSH per gram wet tissue.
Superoxide dismutase (SOD) activity
The SOD activity was measured following the method of Nishikimi et al. using kit purchased from Bio Diagnostic Company. 20 The assay relies on the ability of the SOD enzyme to inhibit the phenazine methosulphate-mediated reduction of nitroblue tetrazolium dye. The SOD activity was expressed as units per gram tissue.
Catalase (CAT) activity
The CAT activity was assayed colorimetrically as described by Sinha using dichromate–acetic acid reagent (5% potassium dichromate and glacial acetic acid were mixed in 1:3 ratio). 21 The intensity was measured at 620 nm, and the amount of hydrogen peroxide hydrolyzed was calculated for the CAT activity.
Measurement of nitrite/nitrate (NOx) content
Gastric mucosa was homogenized in sample buffer containing 10 mM ethylenediaminetetraacetic acid and centrifuged at 13,000× g at 4°C for 5 min. The supernatant was transferred to a new tube and had a total protein content of 10 μg/μL. According to Green et al., the measurement of gastric mucosal NOx was determined. 22 Briefly, NO generated by NOS undergoes a series of reactions and reacts with Griess Reagent to produce a colored product with a strong absorbance atoptical density (OD) 540 nm.
Estimation of inflammatory cytokines
The levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) in gastric supernatants were determined using commercial ELISA kits, according to the manufacturer’s instructions (R&D, Minneapolis, Minnesota, USA).
Immunohistochemical (IHC) analysis
Inducible NO synthetase (iNOS), COX-2, and nuclear factor kappa-B (NF-κB) primary antibodies (Thermo Fisher Scientific, Fremont, California, USA) were used for IHC staining of the stomach sections. Steps of IHC were carried out automatically using Ventana Bench Mark system as previously described. 23 IHC was quantified using image analysis software (Image J, 1.46a, NIH, Bethesda, MD, USA).
Statistical analysis
Statistical analysis was done using one-way analysis of variance, followed by Tukey’s Kramer multiple comparison test. Nonparametric Kruskal–Wallis test followed by Dunn’s multiple comparison posttest was used for analysis of the scores of histopathological lesions. The values are means ± SE. The value of p < 0.05 was considered as significant.
Results
Effect of carvedilol on aspirin-induced histopathological damage of gastric mucosa
As shown in Figure 1, the gastric mucosa obtained from the control animals showed normal histology with intact cellular architecture of the stomach. Administration of aspirin resulted in ulcerative damage of the gastric mucosa. Damaged mucosal epithelium and inflammatory infiltration was observed in the stomachs of the aspirin-treated rats. Treatment with carvedilol concurrently with aspirin remarkably attenuated aspirin-induced gastric damage.
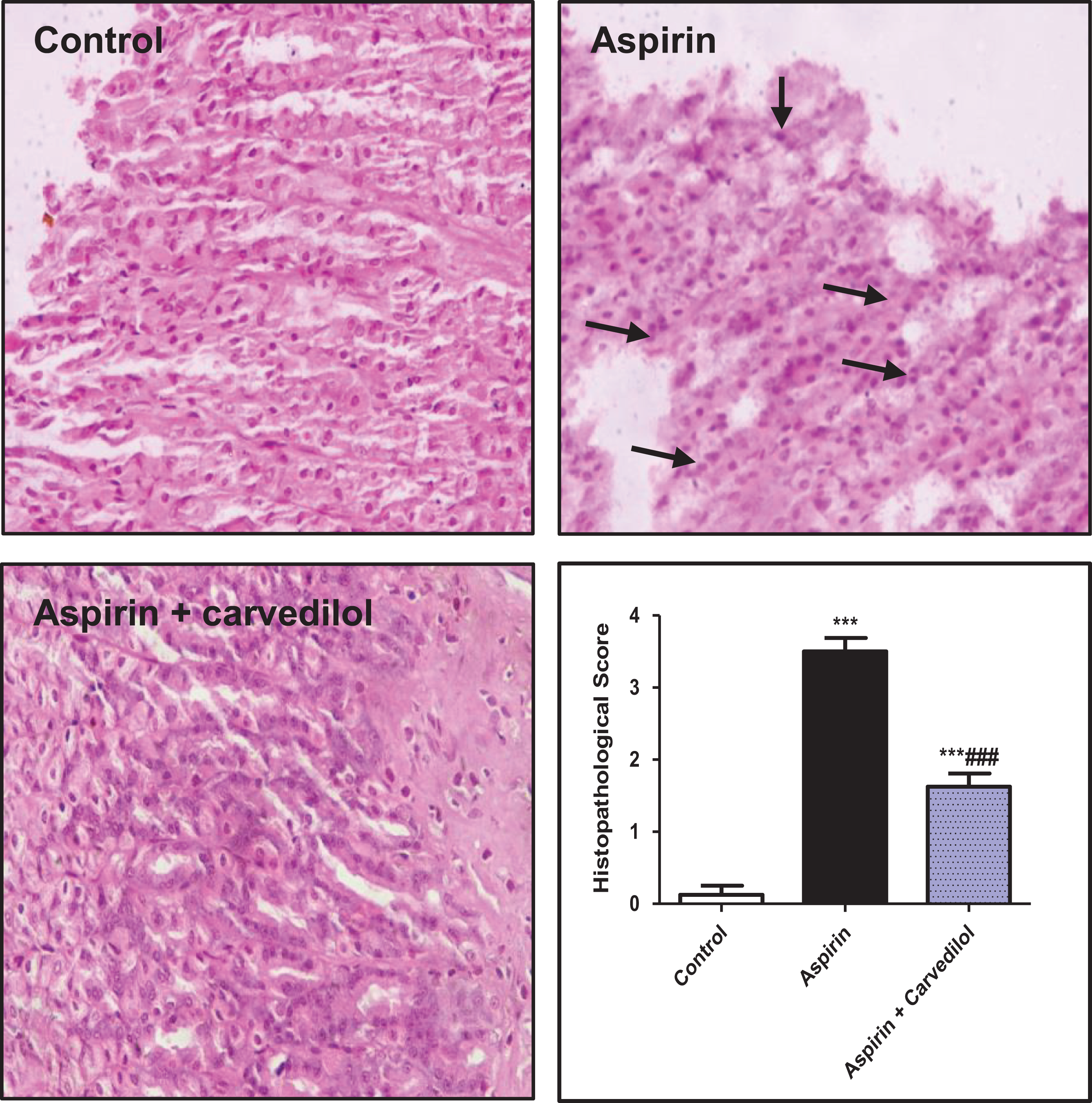
Effect of carvedilol on aspirin-induced histological injury of gastric mucosal tissue. Control group: Gastric tissue is normal with no signs of any lesions. Aspirin group: Ulcerative injury with damaged mucosal epithelium, degeneration, and necrosis is observed. Aspirin + carvedilol group: Gastric mucosa showed a remarkable improvement of the pathological changes (hematoxylin and eosin stain) (400×). Semiquantitative analysis of the gastric lesions. Data are means ± SE (n = 8). ***p < 0.001 compared to the control; ### p < 0.001 compared to the aspirin group (Kruskal–Wallis test followed by Dunn’s multiple comparison).
Effect of carvedilol on oxidative stress and antioxidants
Aspirin administration caused increase in lipid per oxidative products, MDA, 4-HNE, and PC. Furthermore, aspirin depressed the antioxidants, GSH, SOD, and CAT as compared with normal animals (Figure 2). Carvedilol treatment significantly counteracted these changes and restored the normal levels of antioxidants.
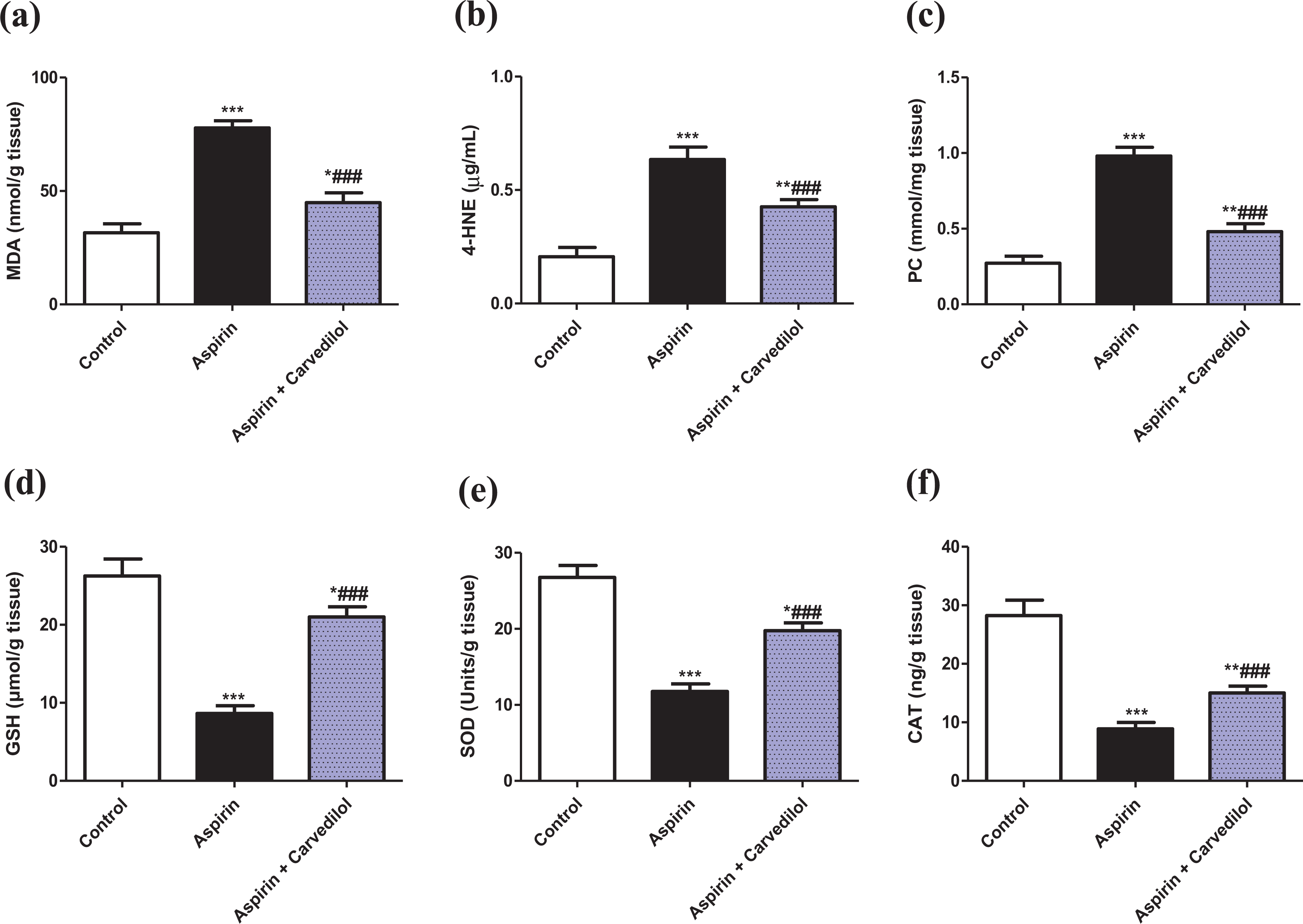
Effect of carvedilol on gastric mucosal lipid peroxidation and antioxidants in aspirin-induced gastric ulcer: (a) MDA, (b) 4-HNE, (c) PC, (d) GSH, (e) SOD, (f) CAT. Data are means ± SE (n = 8). *p < 0.05; **p < 0.01; ***p < 0.001 compared to the control; ### p < 0.001 compared to the aspirin group (one-way ANOVA followed by Tukey–Kramer multiple comparisons test). MDA: malondialdehyde; 4-HNE: 4-hydroxynonenal; PC: protein carbonyl; GSH: reduced glutathione; SOD: Superoxide dismutase; CAT: catalase; ANOVA: analysis of variance.
Effect of carvedilol on COX-2 and NF-κB immunoexpression
Aspirin significantly enhanced the immunoexpression of COX-2 (Figure 3) and NF-κB (Figure 4) as compared to control rats. On the contrary, carvedilol-treated rats exhibited minimal expression of COX-2 and NF-κB.

Effect of carvedilol on aspirin-induced COX-2 immunoexpression in stomach. Representative COX-2 immunostaining of stomach sections (200×). Control group exhibited no COX-2 immunostaining. Aspirin group showed intense brown COX-2 immunostaining (arrows). Aspirin + carvedilol group showed remarkable attenuation in COX-2 immunostaining. Semiquantitative analysis of COX-2 immunostaining in gastric tissue among different groups is expressed as percentage of COX-2 immunopositive cells. Data are means ± SE (n = 8). ***p < 0.001compared to the control; ### p < 0.001 compared to the aspirin group (one-way ANOVA followed by Tukey–Kramer multiple comparisons test). COX-2: cyclooxygenase-2; ANOVA: analysis of variance.
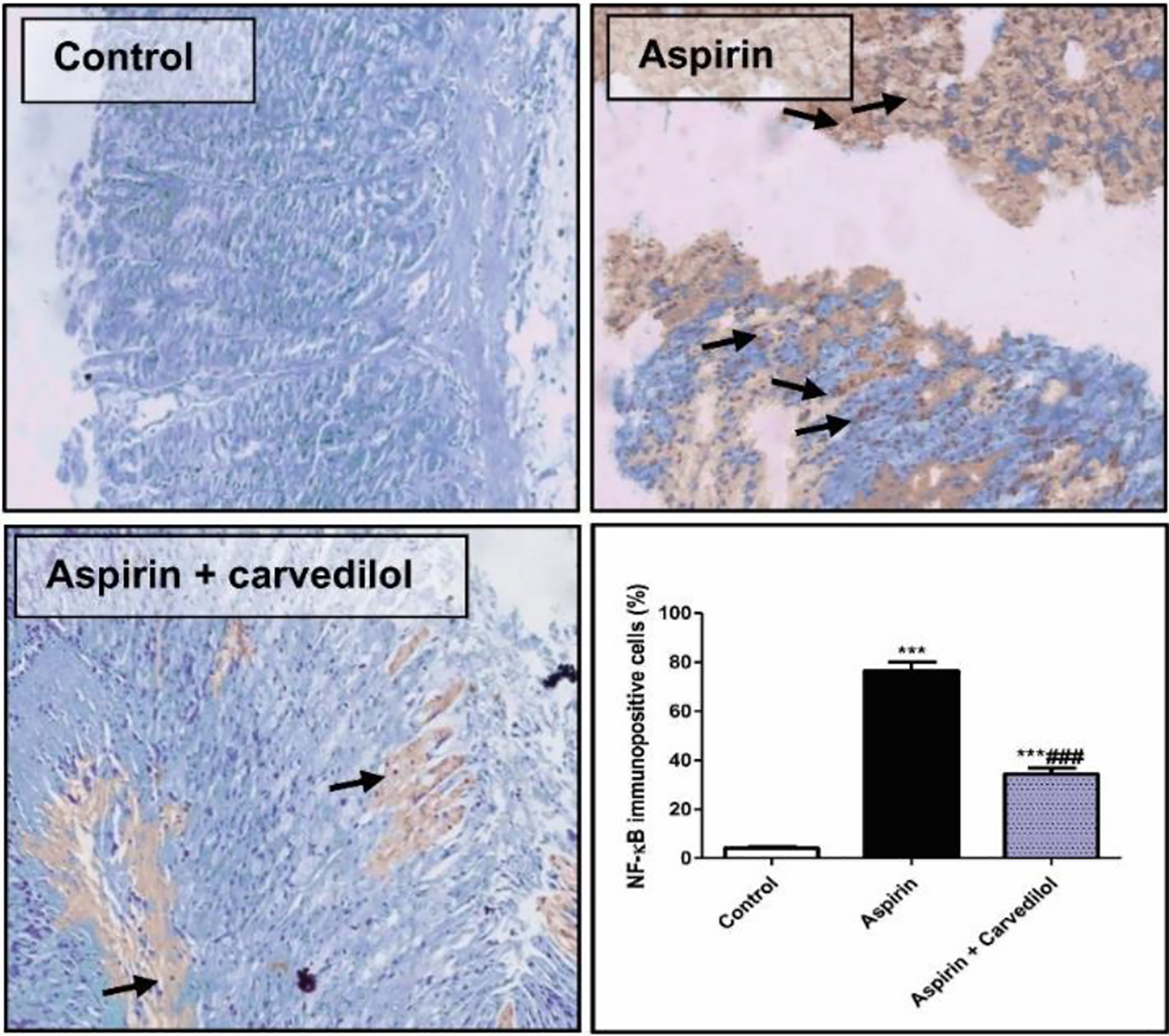
Effect of carvedilol on aspirin-induced NF-κB immunoexpression in stomach. Representative NF-κB immunostaining of stomach sections (200×). Control group had negative immunoexpression of NF-κB. Aspirin group showed intense brown NF-κB immunostaining (arrows). Aspirin + carvedilol group showed a significant reduction in NF-κB immunostaining. Semiquantitative analysis of NF-κB immunostaining in gastric tissue among different groups is expressed as percentage of NF-κB immunopositive cells. Data are means ± SE (n = 8). ***p < 0.001 compared to the control group; ### p < 0.001 compared to the aspirin group (one-way ANOVA followed by Tukey–Kramer multiple comparisons test). NF-κB: nuclear factor kappa-B; ANOVA: analysis of variance.
Effect of carvedilol on cytokine release
As shown in Figure 5, TNF-α, IL-6, and IL-1β were significantly elevated after the administration of aspirin compared to the control animals. On the other hand, carvedilol markedly attenuated these increases in inflammatory mediators.

Effect of carvedilol on aspirin-associated cytokine release: (a) TNF-α, (b) IL-6, (c) IL-1β. Data are means ± SE (n = 8). *p < 0.05; ***p < 0.001 compared to the control group; ### p < 0.001 compared to the aspirin group (one-way ANOVA followed by Tukey–Kramer multiple comparisons test). TNF-α: tumor necrosis factor-α; IL-6: interleukin-6; IL-1β : interleukin-1β; ANOVA: analysis of variance.
Effect of carvedilol on iNOS immunoexpression and NOx level
Aspirin administration resulted in a significant increase in the immunoexpression of iNOS and rise in the level of gastric NOx compared to the control group. However, carvedilol treatment significantly counteracted these changes (Figure 6).

Effect of carvedilol on aspirin-induced alteration in iNOS immunoexpression and NOx level in stomach—(a) representative iNOS immunostaining of stomach sections (200×): control group with no detectable iNOS immunoexpression; aspirin group showed intense brown iNOS immunostaining (arrows); aspirin + carvedilol group exhibited a significant amelioration iNOS immunostaining; semiquantitative analysis of iNOS immunostaining in gastric tissue among different groups is expressed as percentage of iNOS immunopositive cells. (b) NOx level in the supernatants of tissue homogenates. Data are means ± SE (n = 8). ***p < 0.001 compared to the control group; ### p < 0.001 compared to the aspirin group (one-way ANOVA followed by Tukey–Kramer multiple comparisons test). iNOS: nitric oxide synthetase; NOx: nitrite/nitrate; ANOVA: analysis of variance.
Discussion
Gastritis is one of the most common gastrointestinal disorders affecting about 10% of the population. NSAIDs such as aspirin are used for inhibiting inflammation, relieving pain, and also as a prophylactic agent for cardiovascular diseases. 24 Despite its clinical applications, it has some major limitations such as gastric ulcer and bleeding. 25 Previous reports have suggested that the use of aspirin is linked with the development of ulcers. 26 Carvedilol is a known antihypertensive agent which has marked antioxidant activities. Previous studies have discussed the ability of carvedilol to decrease gastric secretion and to protect against ethanol-induced gastric damage. 27,28 Based on these previous data, this study investigated the gastroprotective activity of carvedilol against aspirin-induced gastritis.
Results of the present study revealed that aspirin induced histopathological gastric lesions in the form of congestion, inflammation, hemorrhage, edema, dysplastic changes, and necrosis. A significant improvement was observed in the carvedilol pretreatment animals indicating its gastro-protective activity. These results agree with the former study, which proved the inhibition of ethanol-induced gastric damage with carvedilol pretreatment. 28 To further evaluate the gastro-protective activity of carvedilol, mechanistic studies were addressed focusing on the modulation of oxidative stress and inflammation.
The well-documented mechanism of aspirin-induced gastric damage is inhibition of COX enzyme, which is involved in PG synthesis. PG is one of the defensive factors that protect gastric mucosa, thereby its inhibition can lead to damage. 29 Among the two types of COX isoenzymes, COX-1 possesses mucosal protective role by regulating acid secretion. Investigation showed that NSAIDs should inhibit both the isoforms to possess its ulcer-forming effect. Ironically, COX-2 expression was aggravated with consequent administration of NSAIDs. 30 This investigation is consistent with our results which proved the exacerbation of COX-2 expression with aspirin administration. IHC analysis exhibited that carvedilol administration resulted in the inhibition of COX-2 expression. This finding can be strengthened with previous data that also demonstrated the inhibitory effect of carvedilol on COX-2. 31 The mechanism probably could be due to inhibiting Kuppfer cell activation, thus reducing COX-2 production subsequently.
Oxidative stress plays a pivotal role in deteriorating cell function resulting to various pathological states. 32 The crucial mechanisms of aspirin-induced toxicity are production of free radicals, amelioration of antioxidant enzymes, and increase in the level of MDA-indicating lipid peroxidation. 33,34 The mechanism of oxidative stress induced by NSAIDs could be due to its effect on mitochondria. Uncoupling of oxidative phosphorylation leading to disturbance in the membrane potential is been reported. 35 This could lead to alteration in the permeability of mitochondrial membrane causing release of cytochrome c from inner space into cytosol. Liberated cytochrome c produces ROS such as hydrogen peroxide, consequential activation of caspase 3, caspase 9, and lipid peroxidation resulting in apoptosis. 36 Our study confirmed the role of oxidative stress in aspirin-induced gastritis. There was an increase in the level of MDA and 4-HNE outcoming to lipid peroxidation and PC (protein peroxidative product). In addition, aspirin reduced the levels of antioxidants such as GSH, SOD, and CAT. These results are in accordance with the previous studies. 37 Interestingly, subsequent reduction in the levels of peroxidative markers (MDA, 4-HNE, and PC) and augmentation of antioxidant capacities were observed with carvedilol pretreatment. This antioxidative properties of carvedilol have been reported earlier in many studies. 38 –40 It may be acceptable to link the gastro-protective activity of carvedilol to its antioxidant potential probably by scavenging free radicals.
The molecular mechanism of aspirin-induced gastric damage includes the activation of transcription factor NF-κB. 41 It was shown that aspirin can activate NF-κB resulting to apoptosis in colon neoplastic epithelial cells by phosphorylation and subsequent degradation of IκB, thereby outcoming to the translocation of NF-κB to nucleus where it regulates transcription of inflammatory genes. 42 The present results indicated that aspirin activates NF-κB, a finding that aligned with the above findings. Interestingly, carvedilol administration resulted in the amelioration of NF-κB. This finding can be supported by previous studies that also showed the inhibitory effect of carvedilol on NF-κB, which could be due to its anti-oxidative potential that averts IκB degradation. 43
Aspirin-induced gastric damage involves infiltration of neutrophils and inflammatory response. 44 It has been reported that there is an augmentation of TNF-α release leading to the production of superoxide radicals by neutrophils 45 and activation of IL-1β resulting to the accumulation of neutrophils. 46 A study conducted on healthy volunteers demonstrated that a rebound increase in cytokines after aspirin administration was observed. 47 The present results were in the same direction with earlier ones as there were increased level of TNF-α, IL-1β, and IL-6, following aspirin administration. Carvedilol pretreatment resulted in marked amelioration of inflammatory cytokines release. These findings are in harmony with the previous studies which reported the anti-inflammatory activity of carvedilol. 30,48,49 These data lead us to presume that anti-inflammatory effect of carvedilol against aspirin-induced gastritis may be mediated through inhibition of NF-κB and cytokine release.
Increase in NOx production due to iNOS expression also plays a crucial role in aspirin-induced toxicity. 50 It has been reported that high amount of NO produced from iNOS enzyme causes damage to epithelium. 51 Deleterious effects on the gastric epithelial cells due to aspirin administration were noticed in previous reports. 52,53 The release of excess NOx is ascribed to nitrogen free radical species, which can be formed by the reaction of NO with oxygen and superoxide. 54 Our results showed enhancement of iNOS expression after aspirin administration, which is parallel with the above studies. Furthermore, NOx level was elevated in the gastric tissue. A significant reduction in iNOS expression was observed with carvedilol pretreatment, which can be supported by a previous study that proved the diminishing effect of carvedilol on NO. 49 The suggested anti-inflammatory mechanism of carvedilol may be somewhat linked to the inhibition of NOx production. 55 In conclusion, the gastro-protective effect of carvedilol against aspirin-induced gastric damage was demonstrated. This effect can be attributed to its antioxidant and anti-inflammatory potential, thereby interfering with NF-κB/COX-2/iNOS pathways. These data may be of high clinical relevance as it highlights carvedilol as a potential gastroprotective agent. Further studies are encouraged to get a deeper elucidation of the molecular events underlying these protective effects of carvedilol.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.