
Abstract
Lidocaine, a typical local anesthetic, has been shown to directly induce neurotoxicity in clinical settings. Dexmedetomidine (DEX) is an alpha-2-adrenoreceptor agonist that has been used as anxiolytic, sedative, and analgesic agent which has recently found to protect against lidocaine-induced neurotoxicity. Nicotinamide adenine dinucleotide-dependent deacetylase sirtuin-1 (SIRT1)/forkhead box O3 (FOXO3a) signaling is critical for maintaining neuronal function and regulation of the apoptotic pathway. In the present study, we designed in vitro and in vivo models to investigate the potential effects of lidocaine and DEX on SIRT1 and FOXO3a and to verify whether SIRT1/FOXO3a-mediated regulation of apoptosis is involved in DEX-induced neuroprotective effects against lidocaine. We found that in both PC12 cells and brains of mice, lidocaine decreased SIRT1 level through promoting the degradation of SIRT1 protein. Lidocaine also increased FOXO3a protein level and increased the acetylation of SIRT1 through inhibiting SIRT1. Upregulation of SIRT1 or downregulation of FOXO3a significantly inhibited lidocaine-induced changes in both cell viability and apoptosis. DEX significantly inhibited the lidocaine-induced decrease of SIRT1 protein level and increase of FOXO3a protein level and acetylation of FOXO3a. Downregulation of SIRT1 or upregulation of FOXO3a suppressed DEX-induced neuroprotective effects against lidocaine. The data suggest that SIRT1/FOXO3a is a potential novel target for alleviating lidocaine-induced neurotoxicity and provide more theoretical support for the use of DEX as an effective adjunct to alleviate chronic neurotoxicity induced by lidocaine.
Introduction
Local anesthetics are considered to reduce perioperative pain and the stress after various types of surgery in the clinical settings. 1 However, these local anesthetics, such as lidocaine, have been found to result in neurotoxicity. 2,3 Lidocaine is typically used to reduce perioperative pain, 1 which has been shown to directly induce neurotoxicity through the induction of cell apoptosis. 2,4 –6 The adverse effects of lidocaine have raised concerns about its use. 7 Despite the improved treatment guidelines for neurotoxicity induced by local anesthetics including lidocaine, there are still limitations for these treatments. 8 The underlying molecular mechanism of lidocaine-induced neurotoxicity is still not clear. 9
Dexmedetomidine (DEX) is a highly specific and selective alpha-2(α2)-adrenoreceptor agonist that has been used as anxiolytic, sedative, and analgesic agent in small animals and humans. 10,11 DEX is well known for the ability to provide sedation without inducing respiratory depression. 12 Several studies also suggest that DEX exhibits various effects, such as antioxidant and anti-inflammatory activities. 13,14 DEX can also be used as an adjunct with other sedatives or anesthetics to enhance sedation and analgesia. 15 It is interesting that DEX exhibits a neuroprotective activity via inhibition of apoptosis in brain injury models. 13 In particular, it has been reported that DEX could inhibit lidocaine-induced cytotoxicity. Downregulation of stathmin 1 16 and miR-let-7b-mediated downregulation of COL3A1 17 are involved in the protective effects of DEX against lidocaine-induced cytotoxicity. However, the molecular mechanism underlying the neuroprotective effects of DEX is not completely understood.
Nicotinamide adenine dinucleotide-dependent deacetylase sirtuin-1 (SIRT1)/forkhead box O3 (FOXO3a) signaling plays an important role in apoptosis in the nervous system. 18 In the present study, we designed in vitro and in vivo models to investigate the potential effects of lidocaine and DEX on SIRT1 and FOXO3a and to verify whether SIRT1/FOXO3a-mediated regulation of apoptosis is involved in DEX-induced neuroprotective effects against lidocaine.
Materials and methods
Cell culture and treatment
PC12 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, California, USA) containing 10% heat-inactivated fetal bovine serum (Gibco, Grand Island, New York, USA), 100 U/ml penicillin A, and 100 U/ml streptomycin (Invitrogen) at 37°C in a humidified atmosphere with 5% CO2. P15–20 passage of PC12 cells were used in the present study. Differentiation of PC12 cells was induced by nerve growth factor treatment. PC12 cells were treated with 0.25–1 mM lidocaine or treated with 1 mM lidocaine and different concentrations of DEX (10, 25, and 50 μΜ) for 48 h.
The short hairpin RNA (shRNA) against SIRT1 and FOXO3a and shRNA normal control (NC) (scramble) were purchased from Santa Cruz (Santa Cruz, California, USA). The overexpression vector and pcDNA empty vector were synthesized by Gene Pharma (Shanghai, China). For the transfection, cells were transfected with shRNAs or pcDNAs using Lipofectamine 2000 (Invitrogen, USA). Six hours post-transfection, complete culture medium was replaced.
Cell viability
Cell viability was determined using Cell Counting Kit-8 (CCK-8) (BioTime, Shanghai, China). PC12 cells grown in 96-well plates were treated with 0.25–1 mM lidocaine or treated with 1 mM lidocaine and different concentrations of DEX (10, 25, and 50 μΜ). After the incubation, cells were washed with phosphate-buffered saline and 100 µl fresh medium containing 10 µl CCK-8 solution was added and incubated in the incubator for 1–2 h. Finally, the absorbance at 450 nm was measured using a microplate reader (BioRad, Hercules, CA, USA).
Apoptosis analysis
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit was used to measure apoptosis (Promega, Madison, WI, USA). 2 × 105 cells/well were seeded in a 6-well plate. After the transfection and treatment, cells were washed, harvested, and resuspended in 500 µl binding buffer. Then 10 µl TUNEL solution was added and incubated for 20 min in the dark. Finally, cells were washed and analyzed using a flow cytometer (BD Biosciences, San Jose, California, USA).
Western blot analysis
Total protein was extracted using radioimmunoprecipitation assay (RIPA) lysis buffer (BioTime). The supernatant was collected and protein concentration was quantified using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Protein supernatant was mixed with an equal volume of loading buffer. An equal amount of protein was subjected to sodium dodecyl sulfate -polyacrylamide gel electrophoresis (SDS-PAGE), which was then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, Massachusetts, USA). After blocking with 5% bovine serum albumin, the membranes were immersed with primary antibodies overnight in the cold room. After washing with tris-buffered saline and tween 20 (TBST), the membranes were incubated with the horseradish peroxidase -conjugated secondary antibodies. The protein bands were visualized with an electrochemiluminescence kit (Millipore).
Immunoprecipitation and acetylation detection
After the treatment, total proteins from the cells were extracted and incubated with immunoprecipitation-indicated antibody (1 µg) overnight at 4°C. After that, 30 µl protein A/G plus agarose was added to the mixture followed by a 2-h incubation at 4°C forming an immune complex. After centrifugation at 3000 r/min for 4 min at 4°C, four times washing of protein A/G plus agarose beads were performed using 1 ml lysate each. The protein was mixed with an appropriate protein loading buffer and boiled for 5 min. After another centrifugation at 3000 r/min, the supernatants were collected for Western blot determination.
Animal experiments
The animal experiments were approved by the Institutional Animal Care and Use Committee of Shanxi Provincial People’s Hospital. Twenty-four C57BL6 mice (8–10 weeks old) were randomly divided into 4 groups: control, 1, 2, and 4 mg/kg lidocaine groups. Mice in lidocaine groups received 1, 2, or 4 mg/kg subcutaneous (s.c.) lidocaine. Forty-eight hours after the administration, the mice were euthanized and brain tissues were collected for further determination.
Statistical analyses
The experiments were performed in triplicate and data were analyzed using GraphPad Prism 6.0 software (GraphPad Software Inc., La Jolla, California, USA). Data were expressed as mean ± standard deviation and analyzed by one-way analysis of variance followed by Tukey’s post hoc test. Differences were considered to be significant when p < 0.05.
Results
Lidocaine suppresses SIRT1 protein expression in a degradation-dependent manner
To investigate whether lidocaine induces abnormal expression of SIRT1, leading to lidocaine-induced neurotoxicity, we measured the effect of lidocaine on SIRT1 protein expression using immunoblots. We used differentiated PC12 cells as in vitro models to evaluate the cytotoxicity of lidocaine. As shown in Figure 1, incubation of cells with 0.5 and 1 mM lidocaine for 48 h induced a significant decrease in cell viability. In Figure 2(a), we further showed that SIRT1 protein levels were reduced by 0.5 and 1 mM lidocaine. Moreover, we also treated mice with different doses of lidocaine to evaluate the effect on SIRT1 expression. As shown in Figure 2(b), 2 and 4 mg/kg lidocaine significantly reduced the protein levels of SIRT1 in brain tissues. To examine the mechanism of lidocaine-induced decrease in SIRT1 protein level, we determined the messenger RNA (mRNA) expression of SIRT1 in differentiated PC12 cells treated with lidocaine. As illustrated in Figure 2(c), the mRNA levels of SIRT1 were not significantly affected by lidocaine. To determine whether degradation of SIRT1 was influenced by lidocaine, we used an inhibitor of the proteasome, bortezomib. We showed that bortezomib significantly inhibited the lidocaine-induced decrease in the SIRT1 protein level (Figure 2(d)). These results suggest that lidocaine reduces SIRT1 protein levels through regulation of protein degradation in differentiated PC12 cells.

Lidocaine reduces PC12 cell viability. PC12 cells were treated with 0.25, 0.5, or 1 mM lidocaine for 48 h. Cell viability was determined using the CCK-8 assay kit. *p < 0.05. **p < 0.01. CCK-8: Cell Counting Kit-8.

Lidocaine suppresses SIRT1 protein expression in a degradation-dependent manner. (a), (c), and (d) PC12 cells were treated with 0.25, 0.5, or 1 mM lidocaine for 48 h. (a) SIRT1 protein level was determined using Western blot. (b) Mice were treated with 1, 2, or 4 mg/kg lidocaine. Two days post administration, brain tissues were collected and SIRT1 protein level was determined. (c) mRNA level of SIRT1 in lidocaine-treated PC12 cells was determined using RT-qPCR. (d) PC12 cells were treated with 1 mM lidocaine with or without 10 μM bortezomib, an inhibitor of proteasome, for 48 h. The protein level of SIRT1 was examined. SIRT1: sirtuin-1; mRNA: messenger RNA; RT-qPCR: real-time quantitative polymerase chain reaction.
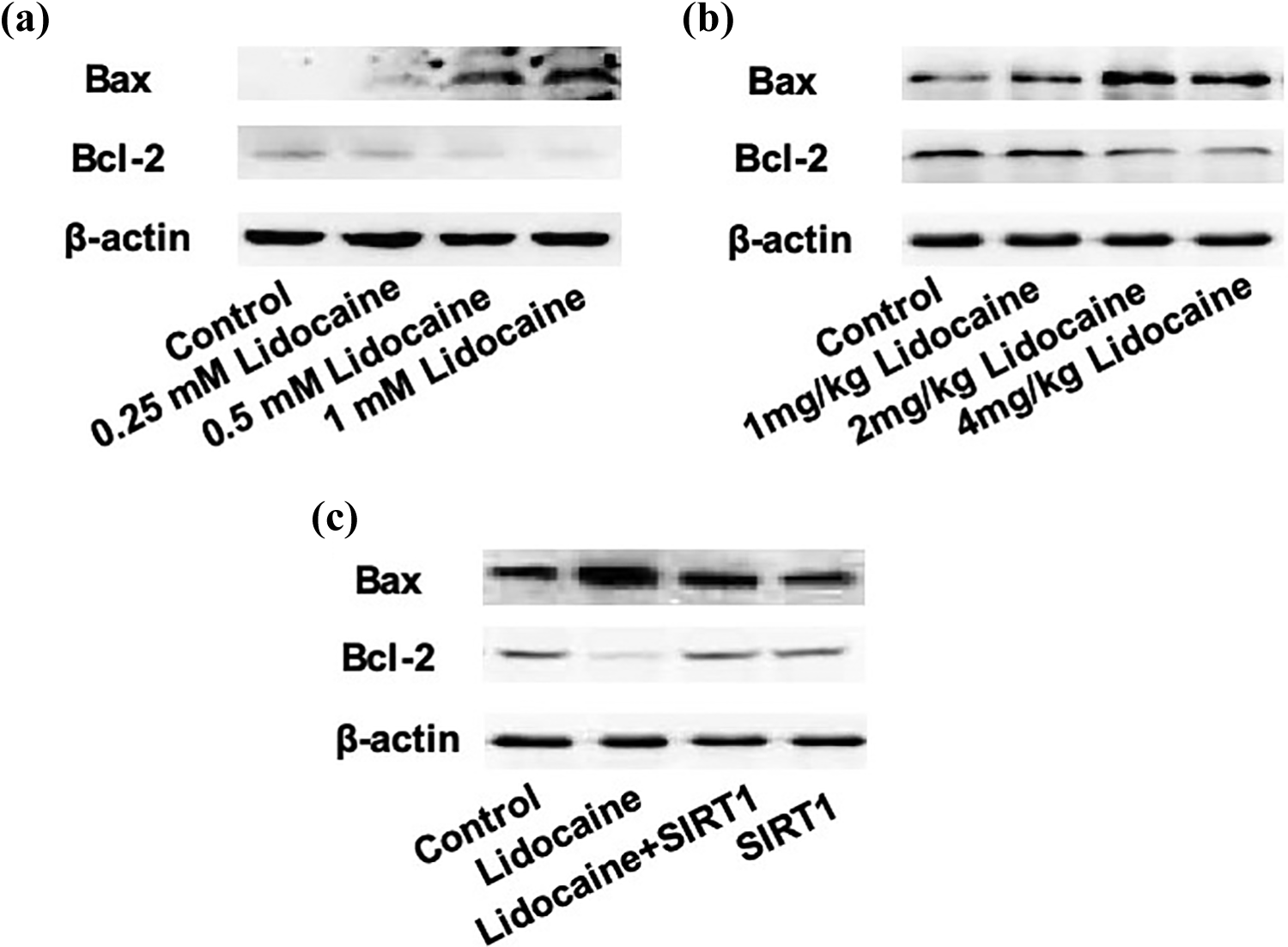
Abnormal expression of SIRT1 is involved in the lidocaine-induced upregulation of Bax and downregulation of Bcl-2. (a) Bax and Bcl-2 protein levels in PC12 cells treated by 0.25, 0.5, or 1 mM lidocaine for 48 h. (b) Bax and Bcl-2 protein levels in brain tissues of mice treated by 1, 2, or 4 mg/kg lidocaine. (c) PC12 cells were transfected with pcDNA-SIRT1 and treated with 1 mM lidocaine for 48 h. (c) Protein levels of Bax and Bcl-2 were examined using Western blot. SIRT1: sirtuin-1.

Abnormal expression of SIRT1 is involved in the lidocaine-induced neurotoxicity. (a) and (b) PC12 cells were transfected with pcDNA-SIRT1 and treated with 1 mM lidocaine for 48 h. (a) Cell viability was determined using CCK-8 assay kit. (b) Apoptosis was detected using TUNEL assay kit. (c) and (d) PC12 cells were treated with 1 mM lidocaine with or without resveratrol for 48 h. (c) Cell viability was determined using CCK-8 assay kit. (d) Apoptosis was detected using TUNEL assay kit. **p < 0.01. ***p < 0.005. SIRT1: sirtuin-1; CCK-8: Cell Counting Kit-8; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Abnormal expression of SIRT1 is involved in lidocaine-induced neurotoxicity
To testify whether abnormal expression of SIRT1 was involved in lidocaine-induced neuronal toxicity, we first examined the effect of lidocaine on apoptotic regulators. As shown in Figure 3(a) and (b), both in PC12 cells and brains of mice, higher doses of lidocaine resulted in a significant increase in BCL2 associated X (Bax) protein levels and decrease in B-cell lymphoma 2 (Bcl-2) protein levels. To confirm the role of abnormal expression of SIRT1 in lidocaine-induced activation of apoptotic signaling, the cells were transfected with pcDNA-SIRT1 and treated with lidocaine. As shown in Figure 3(c), overexpression of SIRT1 significantly inhibited lidocaine-induced increase in Bax and decrease in Bcl-2. Furthermore, the lidocaine-induced decrease in cell viability was inhibited by the overexpression of SIRT1 and the SIRT1 activator, resveratrol (Figure 4(a) and (b)). Lidocaine-induced increase in apoptosis was reduced by the overexpression of SIRT1 and resveratrol (Figure 4(c) and (d)). The results indicated that the reduction in SIRT1 protein levels was involved in lidocaine-induced neurotoxicity.
FOXO3a is activated by lidocaine via inhibition of SIRT1
It is well known that FOXO family members are common downstream targets of SIRT1 and play critical roles in regulating cell viability and apoptotic signaling. In our study, we found that 0.5 and 1 mM lidocaine markedly increased the protein levels of FOXO3a (Figure 5(a)). Moreover, we performed IP using FOXO3a antibody and showed that lidocaine significantly increased acetylation of FOXO3a (Figure 5(b)). To further examine the role of SIRT1 in the lidocaine-induced increase in FOXO3a, we examined the protein level of FOXO3a in lidocaine-treated PC12 cells transfected with pcDNA-SIRT1. The results showed that the overexpression of SIRT1 significantly inhibited lidocaine-induced increase in FOXO3a protein level (Figure 5(c)). Furthermore, we found that the downregulation of FOXO3a inhibited lidocaine-induced decrease in cell viability (Figure 6(a)) and increase in apoptosis (Figure 6(b)). The results demonstrated that lidocaine activated FOXO3a signaling through decreasing SIRT1, which contributed to lidocaine-induced neurotoxicity.
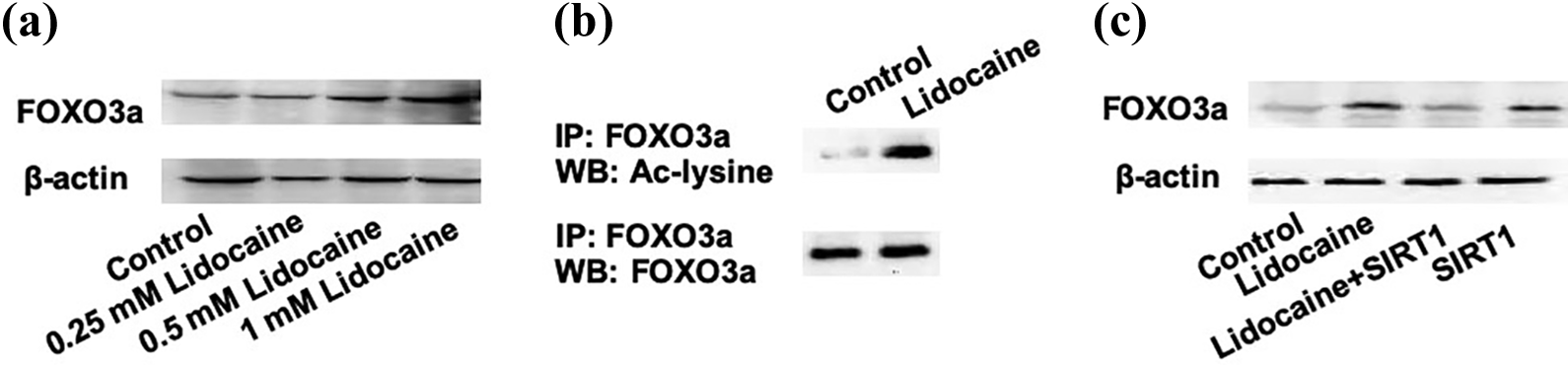
FOXO3a is activated by lidocaine via inhibition of SIRT1. (a) PC12 cells were treated with different concentrations of lidocaine for 48 h and FOXO3a protein level was determined using Western blot. (b) PC12 cells were treated with 1 mM lidocaine for 48 h. IP was performed using FOXO3a antibody and Western blot was performed using FOXO3a and Ac-lysine antibody. (c) PC12 cells were transfected with pcDNA-SIRT1 and treated with 1 mM lidocaine for 48 h. FOXO3a protein level was determined using Western blot. FOXO3a: forkhead box O3; SIRT1: sirtuin-1; IP: immunoprecipitation.
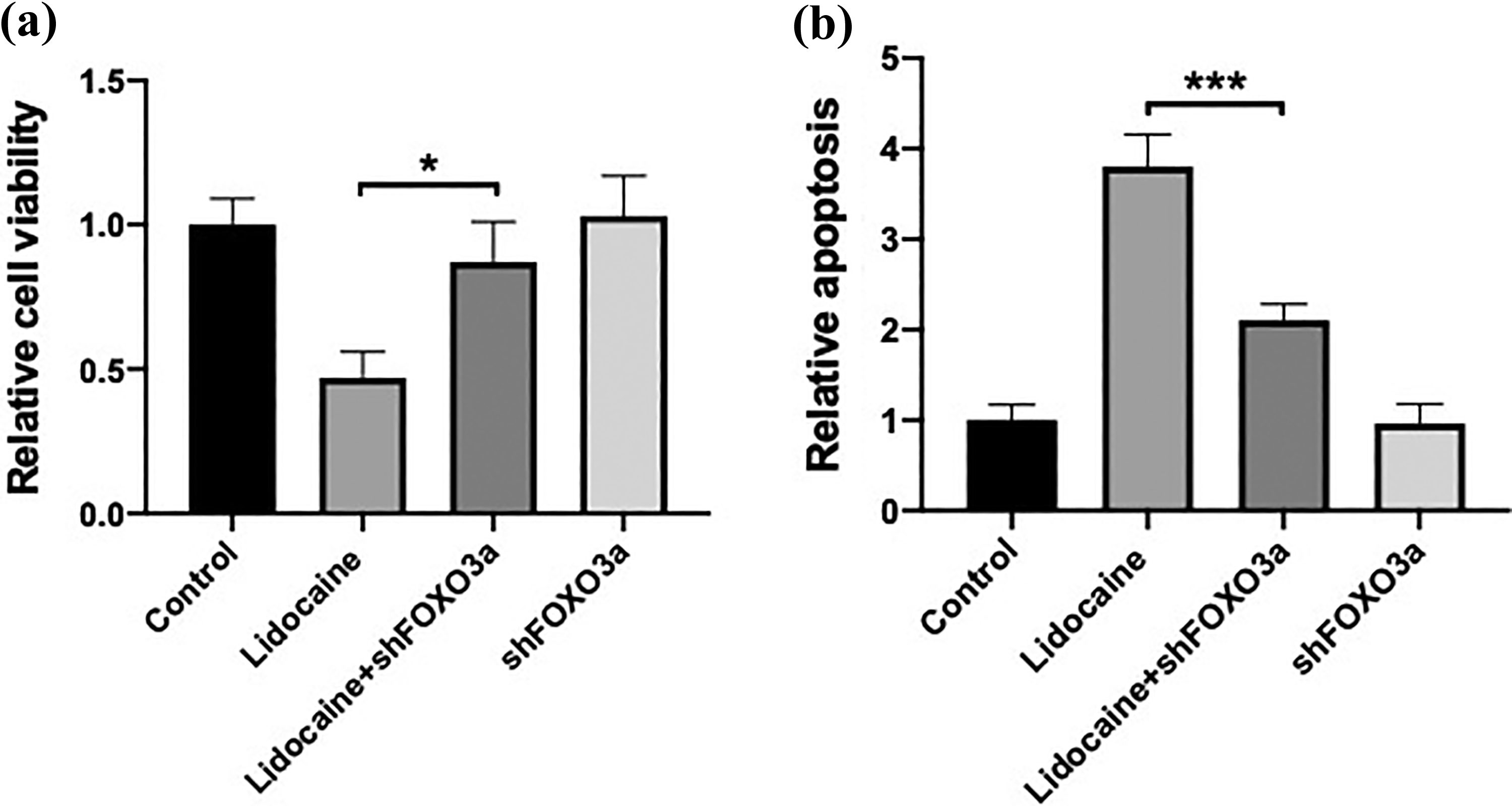
Activation of FOXO3a is involved in the lidocaine-induced neurotoxicity. PC12 cells were transfected with shSIRT1 and treated with 1 mM lidocaine for 48 h. (a) Cell viability was determined using CCK-8 assay kit. (b) Apoptosis was determined using TUNEL assay kit. *p < 0.05. ***p < 0.005. FOXO3a: forkhead box O3; CCK-8: Cell Counting Kit-8; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
DEX inhibits lidocaine-induced cytotoxicity, increases SIRT1, and suppresses FOXO3a
To investigate whether dysregulation of SIRT1 and FOXO3a was involved in the neuroprotective effects of DEX against lidocaine, we examined the protein levels of SIRT1 and FOXO3a. As shown in Figure 7(a), the lidocaine-induced increase in SIRT1 and decrease in FOXO3a were significantly inhibited by DEX, which was in a concentration-dependent manner. Moreover, the lidocaine-induced increase in acetylation of FOXO3a was inhibited by DEX (Figure 7(b)). These findings suggested that DEX inhibited lidocaine-induced downregulation of SIRT1 and activation of FOXO3a. We next determined the effect of DEX on lidocaine-induced cytotoxicity in PC12 cells. As shown in Figure 8(a) and (b), 10–50 μM concentration-dependently increased cell viability and decreased apoptosis in lidocaine-treated PC12 cells. To test the possibility of the involvement of SIRT1/FOXO3a signaling in DEX-induced neuroprotective effects, PC12 cells were transfected with small hairpin RNA (sh) SIRT1 or FOXO3a plasmid and treated by lidocaine and DEX. The results showed that DEX-induced inhibition on lidocaine-induced cytotoxicity was significantly suppressed by the downregulation of SIRT1 or the upregulation of FOXO3a (Figure 8(c) and (d)). This effect was evidenced by decreased cell viability and increased apoptosis in the transfected cells treated by lidocaine and DEX.

DEX inhibits lidocaine-induced increase in FOXO3a and decrease in SIRT1. PC12 cells were treated with 1 mM lidocaine with or without 10, 25, and 50 μM DEX for 48 h. (a) Protein levels of SIRT1 and FOXO3a were examined using Western blot. (b) IP was performed using FOXO3a antibody and Western blot was performed using FOXO3a and Ac-lysine antibody. DEX: dexmedetomidine; FOXO3a: forkhead box O3; SIRT1: sirtuin-1; IP: immunoprecipitation.

DEX inhibits lidocaine-induced cytotoxicity through the regulation of SIRT1 and FOXO3a. (a) and (b) PC12 cells were treated with 1 mM lidocaine with or without 10, 25, and 50 μM DEX for 48 h. (a) Cell viability was determined using CCK-8 assay kit. (b) Apoptosis was determined using TUNEL assay kit. (c) and (d) PC12 cells were transfected with shFOXO3a and treated with 1 mM lidocaine and 50 μM DEX for 48 h. (c) Cell viability was determined using CCK-8 assay kit. (d) Apoptosis was determined using TUNEL assay kit. *p < 0.05. **p < 0.01. ***p < 0.005. DEX: dexmedetomidine; SIRT1: sirtuin-1; FOXO3a: forkhead box O3; CCK-8: Cell Counting Kit-8; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Discussion
Lidocaine is a typical local anesthetic that has been used to reduce perioperative pain and the stress after surgeries. 19 However, the side effect of lidocaine includes neurotoxicity which limits the use of lidocaine. 20,21 Several studies have found that DEX exhibits a neuroprotective effect in the lidocaine-induced model of neurotoxicity. 16,21 –23 However, the mechanism of DEX-related protective effects in lidocaine-induced neurotoxicity is still not completely understood.
In the present study, we used in vitro and in vivo models to investigate the mechanism of lidocaine-induced neurotoxicity and DEX-induced protective effects. The dose of in vivo single injection of lidocaine (4 mg/kg s.c.) was selected according to a previous study. 24 In infants and children, the maximal dose of lidocaine that is allowed for a regional nerve block is 5 mg/kg. 24 Additionally, the median convulsive dose of lidocaine is 289.4 ± 13.4 mg/kg s.c. in mice, 25 suggesting that 4 mg/kg s.c. is comparable to human dose and is a safe dose in mouse.
Considering the critical role of SIRT1 and FOXO3a signaling in the regulation of apoptosis, we tested the possibility of the involvement of SIRT1 and FOXO3a signaling in lidocaine-induced neuronal injury and DEX-induced protective effects. As expected, we found that in both in vitro cultured PC12 cells and in vivo mice models, lidocaine induced a dose-dependent decrease in SIRT1 protein level but not mRNA level. The results suggest that lidocaine reduces SIRT1 protein level through a post-transcriptional mechanism. Proteasome-mediated degradation of SIRT1 has been reported in several disease models. 26 –28 In the present study, we found that the use of a proteasome inhibitor could significantly suppress lidocaine-induced decrease in SIRT1 protein level. Moreover, we transfected the cells with SIRT1 plasmid and treated cells with SIRT1 activator, resveratrol. We revealed that genetic and chemical-induced upregulation of SIRT1 could significantly inhibit lidocaine-induced decrease in cell viability and increase in apoptosis. The results demonstrated that the downregulation of SIRT1 is involved in lidocaine-induced apoptosis in PC12 cells.
We have further shown that FOXO3a protein level was increased by lidocaine. Moreover, acetylation of FOXO3a was increased by lidocaine. Upregulation of SIRT1 notably inhibited lidocaine-induced increase in FOXO3a, indicating that the downregulation of SIRT1 was involved in the lidocaine-induced activation of FOXO3a. SIRT1-mediated regulation of FOXO3a has been found in several different physiological and pathophysiological models. 29,30 Further experiments were performed to verify whether the upregulation of FOXO3a was involved in lidocaine-induced neurotoxicity. Since the downregulation of FOXO3a prevented lidocaine-induced decrease in cell viability and increase in apoptosis, we concluded that the downregulation of FOXO3a was involved in lidocaine-induced neurotoxicity.
Previous studies have suggested the neuroprotective effects of DEX in various models. 31 –34 In recent years, researchers found that DEX inhibited lidocaine-induced neurotoxicity. 16,17,23 In this study, we also verified the role of SIRT1/FOXO3a signaling in the effect of DEX on lidocaine-induced neurotoxicity. We found that DEX concentration-dependently inhibited lidocaine-induced decrease in SIRT1 and increase in FOXO3a and suppressed the increase in acetylation of FOXO3a mediated by reducing SIRT1. Furthermore, DEX-exhibited neuroprotective effects against lidocaine were inhibited by the downregulation of SIRT1 and upregulation of FOXO3a. In a recent study, Chen et al. found that DEX could upregulate SIRT1 and protect against diabetic hyperglycemia-exacerbated cerebral ischemia/reperfusion injury. 35 Consistent with this finding, our data suggest that SIRT1 is a probable target of DEX which mediates its protective effects.
Lidocaine and DEX are two different kinds of drugs. Voltage-gated Na+ channels are reported to the major targets of lidocaine in the neuronal and cardiac cell membrane. 36,37 It is reported that SIRT1 can regulate cardiac sodium channels by deacetylating. 38 Our present study indicates that lidocaine may regulate SIRT1 through sodium channels. The interaction between SIRT1 and sodium channels increases the complexity of the pharmacological effects of lidocaine. DEX mainly functions through two receptors, including α2-adrenergic receptor and imidazoline I2 receptor. 39 It has been found that DEX could inhibit voltage-gated sodium channels via α2-adrenoceptors in trigeminal ganglion neurons. 40 It is suggested that DEX activates its receptors and inhibits sodium channels induced by lidocaine, resulting in the reverse of inactivated SIRT1 and leading to inhibition of apoptosis. Further studies are needed to verify the interaction between DEX receptors, lidocaine receptor, and SIRT1/FOXO3a signaling (Figure 9).

Proposed schematic figure of DEX-induced protective effects against lidocaine-induced neurotoxicity. DEX: dexmedetomidine.
In conclusion, we identified that downregulation of SIRT1 protein and acetylation-mediated activation of FOXO3a were involved in lidocaine-induced neurotoxicity. DEX could upregulate SIRT1 and inhibit FOXO3a, leading to the inhibition of lidocaine-induced neurotoxicity (Figure 9). The data suggest that SIRT1/FOXO3a is a potential and novel target for the alleviation of lidocaine-induced neurotoxicity and provide more theoretical support for the use of DEX as an effective adjunct to alleviate chronic neurotoxicity induced by lidocaine.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Shanxi Provincial People’s Hospital.