
Abstract
Background
Acute lymphoblastic leukemia (ALL) is one of the most common pediatric cancers, characterized by the malignant proliferation of leukemic cells. Despite advancements in treatment, the prognosis for refractory and relapsed ALL remains poor, underscoring the need for novel therapeutic targets and approaches.
Methods
To investigate the anti-leukemic properties of MG132, MTS assays were employed to assess cell viability, and flow cytometry was used to evaluate apoptosis. Mechanistic studies, including qRT-PCR, Western blotting, and lentivirus-mediated FOXO3a knockdown, were conducted to explore MG132’s effects on the Akt/FOXO3a/Bim signaling pathway. A xenograft mouse model was utilized to validate the in vivo efficacy of MG132 in suppressing tumor growth.
Results
MG132 inhibited cell proliferation and induced apoptosis in both ALL cell lines and primary cells in a concentration-dependent manner. Mechanistic studies revealed that MG132 promoted FOXO3a nuclear localization by suppressing Akt phosphorylation and preventing FOXO3a degradation, leading to increased Bim expression. Furthermore, FOXO3a knockdown significantly reduced MG132’s anti-proliferative effects. In vivo, MG132 markedly inhibited tumor growth in the xenograft model.
Conclusion
These findings suggest that MG132 exerts potent anti-leukemic effects through modulation of the Akt/FOXO3a/Bim axis, offering a promising therapeutic avenue for treating ALL.
Introduction
Acute lymphoblastic leukemia (ALL) is among the most prevalent pediatric cancer. 1 Although recent advancements in the treatment of childhood ALL have led to significant improvements, the outcomes for patients who do not achieve long-term disease-free survival (DFS) or who experience relapse remain suboptimal. 2 Given the current inadequate response to treatment in cases of refractory leukemia, it is imperative to investigate the pathogenesis of refractory and relapsed ALL and to identify novel therapeutic targets for leukemia treatment.
Over the past few years, proteasome inhibitors have emerged as significant chemotherapeutic agents for various malignancies.3,4 MG132, a tri-peptidyl aldehyde and 26S proteasome inhibitor, functions by blocking protein degradation through the ubiquitin-proteasome pathway. 5 This action disrupts the degradation of key regulatory proteins such as NF-κB, caspases, and other apoptosis-related proteins, leading to the suppression of cellular proliferation and the promotion of apoptosis. 6 Numerous studies have confirmed that MG132 can induce apoptosis in multiple myeloma, prostate cancer, ovarian cancer, and other cancer cell types. 7 Moreover, research has demonstrated that MG132 can inhibit cellular proliferation and induce apoptosis in various leukemia cells.8,9 For instance, Sun et al. treated HL60 cells (a myeloid leukemia cell line) with MG132, leading to G2/M phase arrest and subsequent apoptosis, with findings suggesting that the accumulation of p21 protein plays a role in this process. 10 Similarly, Ortiz-Lazareno et al. showed that MG132 increased the apoptosis rate in U937 cells (another myeloid leukemia cell line) by inhibiting the loss of NF-κB and mitochondrial membrane potential (MMP), thereby enhancing sensitivity to doxorubicin. Their study found that the combination of MG132 and doxorubicin induced the up-regulation of apoptosis-related genes such as BAX, DIABLO, NOXA, DR4, and FAS, while down-regulating anti-apoptotic factors like BCL-XL and SURVIVIN. 11 In another study, Bravo-Cuellar et al. treated U937 cells with pentoxifylline and MG132, observing significant changes in apoptotic markers, including cytochrome C release, caspase-3, -8, and -9 activation, and loss of MMP. These results were accompanied by decreased phosphorylation of p65 and reductions in Bcl-2 and Bcl-XL levels, along with the overexpression of DIABLO, BAX, and FAS. 12 However, despite these insights, the specific mechanisms by which MG132 acts on ALL cells, particularly drug-resistant ALL cells, remain poorly understood.
In this study, we investigated the anti-leukemic activity of MG132 in ALL cell lines, primary cells, and in a mouse model. Additionally, we explored the role of MG132 in inducing the nuclear translocation of FOXO3a and its impact on the Akt/FOXO3a/Bim pathway. Collectively, our findings suggest that MG132 could serve as a promising candidate for anti-leukemic treatment by targeting FOXO3a degradation and Akt phosphorylation to stimulate Bim expression.
Materials and methods
Cell lines and culture
Detailed information of involved ALL cell lines.

IC50: half inhibitory concentration.
Bone marrow primary cells
Biological and clinical characteristics of involved patients.

Reagent and mouse model
MG132 (C26H41N3O5, molecular weight 475.62, 99% purity) was purchased from Abcam (UK) and dissolved in DMSO for use. Female BALB/c athymic nude mice, aged 5 weeks, were housed in the pathogen-free environment of the Laboratory Animal Center at Sun Yat-sen University.
MTS assay
The antiproliferative effect of MG132 was assessed using the MTS assay. Cells in the logarithmic growth phase were harvested and seeded into 96-well plates. After 24 h of incubation, various concentrations of MG132 were added to each well (100 µl per well). Control groups were set up by adding equal amounts of culture medium and cells without MG132, while blank control wells contained only culture medium without cells. Under sterile conditions, 20 µl of MTS solution was added to each well after 24 h, followed by incubation at 37°C for 1-3 h. The optical density (OD) was measured at 490 nm using a microplate reader. The experiment was repeated three times, and the average OD value was taken as the final result.
Apoptosis assay
A total of 1–5 × 105 cells were collected and centrifuged at 1000 rpm for 5 min. After discarding the culture medium, the cells were washed with PBS and centrifuged again under the same conditions. The cell pellet was then resuspended in 100 μL of Annexin V solution and incubated in the dark at room temperature for 15 min. Following this, propidium iodide (PI) solution was added, and the samples were incubated for an additional 20 min at 4°C. Finally, the cells were washed again with PBS and then analyzed using flow cytometry.
SDS-PAGE and western blotting
Cells were lysed in buffer containing phenylmethanesulfonylfluoride (PMSF) (Beyotime, Guangzhou, China) and heated at 95°C for 5 min. Equal amounts of protein were resolved using 10% SDS-PAGE and transferred onto methanol-activated PVDF membranes. After blocking with 5% non-fat milk for 1 h, the membranes were incubated overnight at 4°C with primary antibodies (FOXO3a, Akt, p-Akt, Bim, GAPDH, H3). The next day, the membranes were incubated with HRP-conjugated secondary antibodies for 1 h. Protein bands were detected using ECL and exposure times were adjusted as necessary to obtain optimal results using a chemiluminescence imaging system.
RNA extraction and RT-PCR
Primer sequence.

Lentivirus packaging and infection
To knock down FOXO3a in cells, FOXO3a shRNA (Clontech, CA, USA) was co-transfected with three packaging plasmids (pLP1, pLP2, and pLP/VSVG) into 293T cells. The supernatant was harvested after 36 h, and Lenti-Concentrin Virus Precipitation Solution was added to concentrate the lentivirus. After determining the virus titer, MOLT-4 and RS4-11 cell lines were transduced with the viral supernatants and selected using 1 µg/ml puromycin (Sigma).
Tumor xenograft experiment
12 female SPF BALB/c nude mice were obtained from Skyley Co. Ltd (Shanghai Animal Laboratory Center, Chinese Academy of Sciences). After a 3-day acclimation period, the mice were injected with a suspension of CEM-C7 cells, which were in the logarithmic growth phase. A total of 100 µL of this cell suspension (1 × 107 cells) was mixed with an equal volume of Matrigel and implanted subcutaneously in the right axilla of each mouse. Tumors were measured with calipers every other day, with volume calculated using the formula: V = (length × width2)/2. Mice in the control group received 0.2 mL of PBS solution containing 0.1% DMSO, while those in the MG132 group were treated with 0.2 mL of MG132 at a dose of 10 mg/kg daily for 7 days (D1–D7). On day 19, the mice were euthanized via cervical dislocation, and the axillary tumors were excised, weighed, and compared. All animal studies were conducted following the approval of the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Sun Yat-sen University.
Statistical methods
Statistical analyses were performed using SPSS 13.0 software. Data are presented as mean ± standard deviation (SD) derived from three independent experiments. Student’s t test was used to assess differences between the experimental and control groups, with a significance level set at p < .05.
Results
MG132 suppresses cell proliferation in three cell lines
The molecular structure of MG132 is illustrated in Figure 1(A). To evaluate the effect of MG132 on the proliferation of ALL cells, cell viability assays were performed across three ALL cell lines. As shown in Figure 1(B), treatment with increasing concentrations of MG132 resulted in a significant, dose-dependent reduction in cell proliferation in the CEM-C7, MOLT-4, and RS4-11 cell lines. MG132 inhibited cell proliferation in three cell lines. (A) Molecular structure of MG132. (B) CEM-C7, MOLT-4 and RS4-11 cell lines were treated with varying concentrations of MG132 for 24 h. Cell proliferation was assessed using the MTS assay. Results are expressed as mean ± standard deviation from three independent experiments. Statistical significance is indicated by *p < .05 and **p < .01.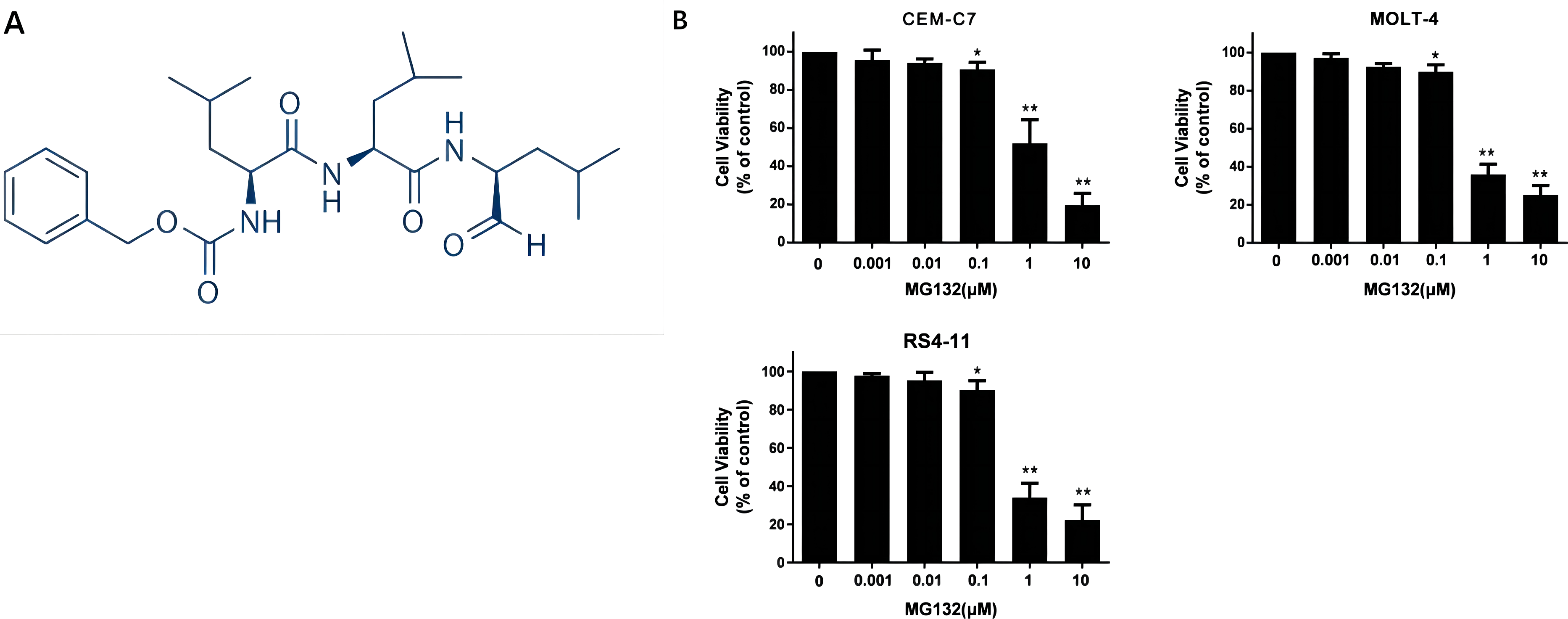
MG132 inhibits the proliferation of primary cells from patients
To assess the anti-proliferative activity of MG132 on primary cells from patients, we conducted MTS assays on both patient-derived cells and normal mononuclear cells from healthy donors. Cells were treated with varying concentrations of MG132 (0, 0.001, 0.01, 0.1, 1, and 10 μM) for 24 h. As shown in Figure 2(A), MG132 significantly inhibited the proliferation of both B-ALL and T-ALL primary cells in a dose-dependent manner. In contrast, at equivalent concentrations, MG132 exhibited minimal cytotoxicity toward normal mononuclear cells (Figure 2(B)), highlighting its selective activity against leukemia cells. MG132 inhibited the proliferation of primary cells from patients in vitro. Cells from five patient samples (A) and five healthy donors (B) were treated with 0, 0.001, 0.01, 0.1, one or 10 μM MG132. MTS assay was used to measure the cell viability.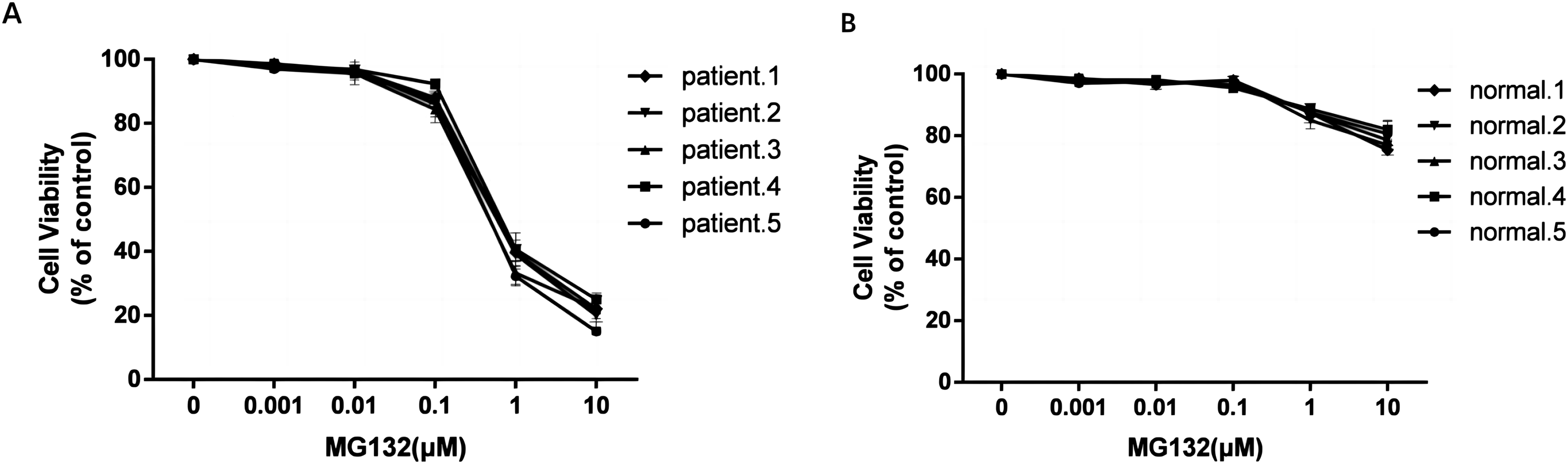
MG132 induces apoptosis in ALL cell lines
To further evaluate the pro-apoptotic effects of MG132, we analyzed apoptosis rates in ALL cell lines treated with increasing concentrations of MG132. In CEM-C7 cells, MG132 treatment at concentrations of 0.5, 1, and 2 μM resulted in a significant, dose-dependent increase in apoptosis (Figure 3(A)). Similarly, both MOLT-4 and RS4-11 cells exhibited markedly elevated apoptotic rates when exposed to 0.25, 0.5, and 1 μM MG132 (Figure 3(B) and (C)). These results demonstrate the potent ability of MG132 to induce apoptosis in multiple ALL cell lines. MG132 induced cell apoptosis in three cell lines. (A) CEM-C7 cells were treated with 0.5, 1, and 2 μM MG132 for 24 h (B) MOLT-4 cells were treated with 0.25, 0.5, and 1 μM MG132 for 24 h (C) RS4-11 cells were treated with 0.25, 0.5, and 1 μM MG132 for 24 h. Cell apoptosis was assessed using flow cytometric analysis. **p < .01 versus control group.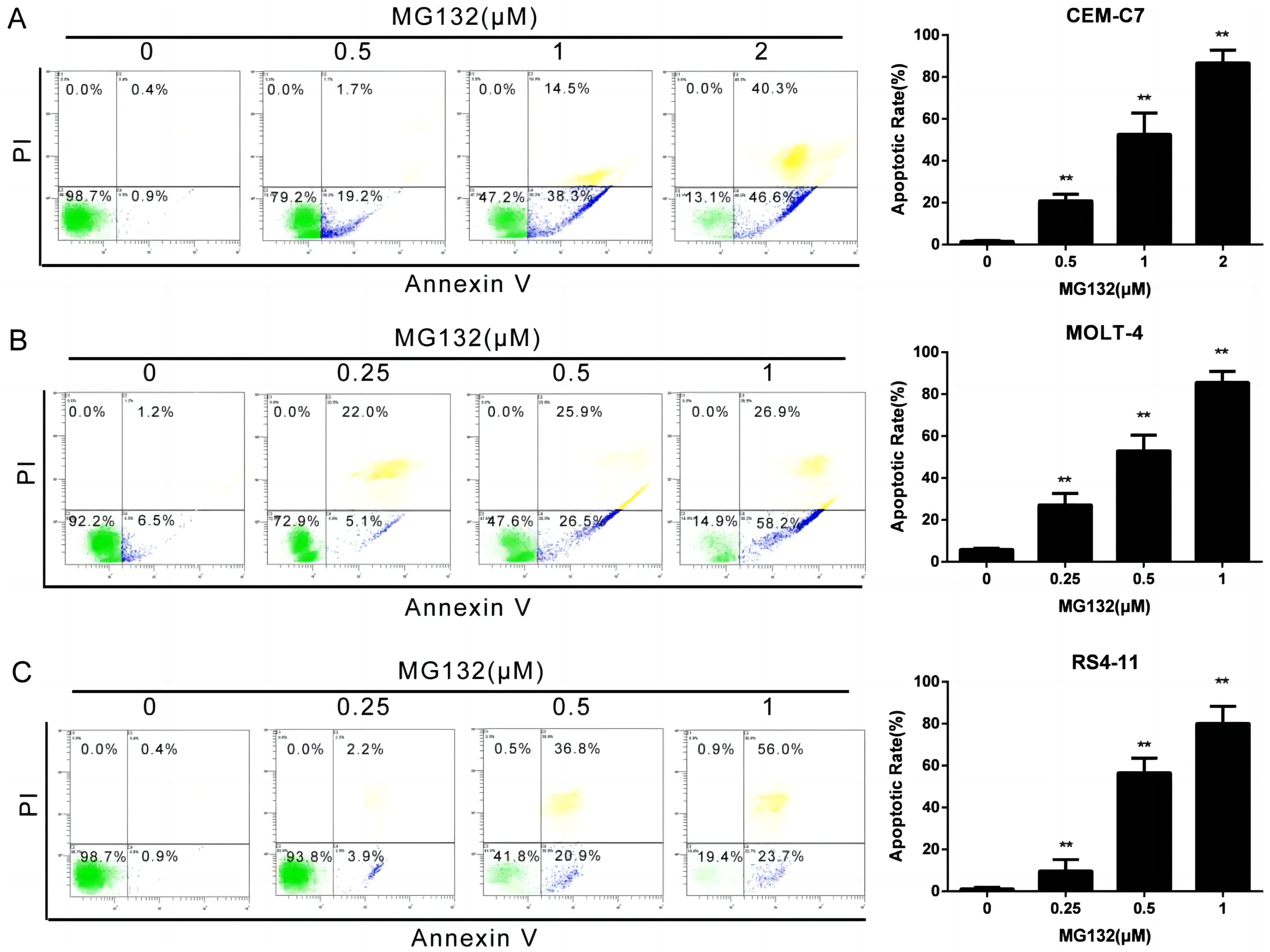
MG132 activates the Akt/FOXO3a/Bim pathway
To determine whether MG132 influences the Akt/FOXO3a/Bim signaling pathway, we assessed the expression levels of key components in ALL cell lines. As shown in Figure 4(A), treatment with MG132 did not notably upregulate the mRNA expression of FOXO3a, though it significantly increased Bim expression. Interestingly, at the protein level, MG132 markedly elevated the expression of both FOXO3a and Bim (Figure 4(B)). Additionally, MG132 treatment led to a significant reduction in the protein levels of Akt and p-Akt, while the levels of phosphorylated FOXO3a (P-FOXO3a) remained unchanged. MG132 activated Akt/FOXO3a/Bim pathway. (A) qRT-PCR was performed to assess the relative mRNA levels of FOXO3a and Bim. (B) ALL cell lines were treated with either 0 or 0.5 μM MG132 for 12 or 24 h, followed by immunoblotting to detect protein expression. (C) Cells were treated with 0 or 0.5 μM MG132 for 12 and 24 h, and immunoblotting with anti-FOXO3a was conducted to analyze protein levels in each cellular fraction. Histone H3 served as an internal control for the nuclear fraction, while GAPDH served as the control for the cytoplasmic fraction.
We further evaluated the impact of MG132 on FOXO3a localization using Western blot analysis. The results indicated a substantial increase in FOXO3a protein within the nucleus compared to the control group, while a slight increase in FOXO3a expression was observed in the cytoplasm (Figure 4(C)).
FOXO3a knockdown attenuates the antiproliferative effect of MG132
To further elucidate the mechanism by which MG132 enhances FOXO3a and Bim expression, we knocked down FOXO3a using two distinct lentiviral shRNAs (shRNA1 and shRNA2) in MOLT-4 and RS4-11 cells. The knockdown of FOXO3a led to a significant reduction in both FOXO3a and Bim mRNA levels (Figure 5(A)). Similarly, the protein expression levels of FOXO3a and Bim were decreased following FOXO3a knockdown (Figure 5(B)). Importantly, knockdown of FOXO3a attenuated the antiproliferative effect of MG132 across different concentrations (Figure 5(C)). FOXO3a knockdown attenuated the antiproliferative effect of MG132. (A) The mRNA levels of FOXO3a and Bim were assessed following knockdown using qRT-PCR. (B) Western blotting was performed to determine the protein levels of FOXO3a and Bim. (C) Cell viability of FOXO3a knockdown and control cells was analyzed using the MTS assay at various concentrations of MG132.
MG132 inhibits tumor growth in ALL mouse models
To further assess the antiproliferative effect of MG132, we established an ALL xenograft mouse model by subcutaneously injecting CEM-C7 cells into nude mice. Mice were subsequently treated with MG132 via intraperitoneal injection every other day for 3 weeks. As demonstrated by the tumor growth curve (Figure 6(A)), MG132 treatment significantly inhibited tumor growth compared to the control group. At the end of the experiment (day 19), tumors were excised and weighed. MG132-treated mice exhibited a marked reduction in both tumor volume and weight (Figure 6(B)), confirming the in vivo efficacy of MG132 in suppressing ALL tumor progression. MG132 inhibited tumor growth in ALL mouse models. (A) Changes in subcutaneous tumor volume were monitored in nude mice treated with MG132 compared to the control group. (B) Comparison of subcutaneous tumor volume and weight between the MG132 and control groups. *p < .05 indicates statistical significance.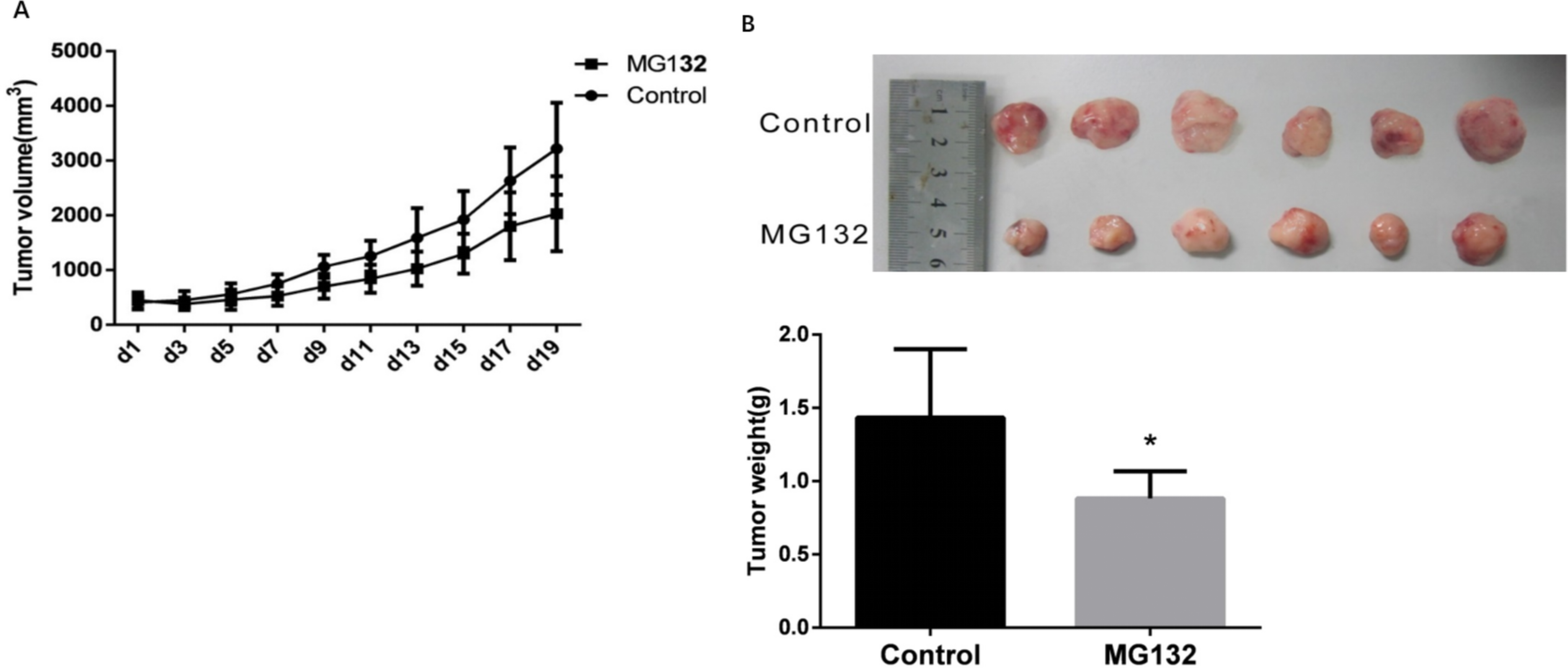
Discussion
ALL is one of the most prevalent pediatric hematological cancers, and modern therapeutic approaches have significantly improved patient outcomes.13,14 However, the treatment of refractory or relapsed ALL continues to pose a significant challenge. 15 Factors such as the recurrence time, location, and immunophenotypic characteristics after initial therapy play a crucial role in determining the prognosis of relapsed pediatric ALL.16,17 This highlights the need for further investigation into the pathogenesis of refractory and relapsed ALL and the identification of novel therapeutic targets to overcome resistance to conventional treatments.
Previous researches have linked FOXO3a phosphorylation to poor prognosis in acute myeloid leukemia (AML). Hypomethylating agents have been shown to dephosphorylate FOXO3a, promoting its nuclear translocation and upregulating pro-apoptotic genes such as Bim and PUMA, thereby inducing apoptosis. 18 In our study, we identified FOXO3a as a key target of MG132. Treatment with MG132 significantly increased FOXO3a protein levels in ALL cell lines, and FOXO3a knockdown reduced the sensitivity of these cells to MG132, highlighting the critical role of FOXO3a in MG132-induced apoptosis. These findings align with previous research by Dewar et al., 19 who successfully used bortezomib, a proteasome inhibitor similar to MG132, to treat a patient with Philadelphia chromosome-positive (Ph+) ALL unresponsive to conventional therapies (imatinib + chemotherapy). This suggests that MG132, like bortezomib, could hold potential for clinical use in refractory ALL.
Additionally, we observed an increase in FOXO3a protein levels following MG132 treatment in ALL cells, while qRT-PCR analysis indicated no significant change in FOXO3a mRNA expression. This suggests that the rise in FOXO3a protein levels is primarily attributed to reduced degradation rather than an increase in transcription. This finding aligns with the established understanding of FOXO3a degradation through the UPP pathway. 20 Moreover, we noted a significant accumulation of FOXO3a within the nucleus. To investigate this further, we examined Akt, a key upstream regulator of FOXO3a, and found a marked decrease in both Akt and p-Akt protein levels after MG132 treatment. Akt is crucial for regulating the phosphorylation and cellular localization of FOXO3a. Therefore, we hypothesize that MG132 not only suppresses Akt expression but also reduces FOXO3a phosphorylation, facilitating its nuclear translocation.
Furthermore, our study revealed a significant upregulation of Bim at both mRNA and protein levels following MG132 treatment. Moreover, WB analysis indicated that the expression of FOXO3a and Bim proteins decreased after FOXO3a shRNA interference. These findings suggest that Bim functions as a downstream target gene of FOXO3a in ALL cells. Previous studies have established that Bim is a member of the pro-apoptotic Bcl-2 family, known for its role in antagonizing cell survival, activating caspases, and promoting apoptosis by interacting with other family members.21,22 Bim is recognized for its critical role in tumor suppression and apoptosis induction across various cancer types.23,24 The involvement of the FOXO3a/Bim pathway in cancer cell lines has been well-documented. Upregulation of FOXO3a can activate Bim expression, thereby inducing apoptosis in tumor cells.25–28
Additionally, it is noteworthy that both the cell lines selected for this study and the primary cells obtained from patients contained both GC sensitive and resistant populations. The experimental results indicated that MG132 was effective against both types, suggesting its potential as a promising therapeutic target for treating clinically drug-resistant ALL patients. Junk et al. 29 found that bortezomib could reverse the GC resistance in ALL, attributing this effect to the regulation of proteins associated with the ubiquitin-proteasome degradation system; however, the specific mechanisms underlying this phenomenon remain inadequately explored. Given that MG132 is also a proteasome inhibitor, investigating whether it can enhance the sensitivity of ALL cells to glucocorticoids, along with the underlying mechanisms, warrants further exploration. Nevertheless, this study has some limitations. For instance, we did not investigate other upstream regulators of FOXO3a, such as IKKβ, ERK, and various phosphorylation signals, nor did we validate the FOXO3a signaling pathway in vivo. Future studies will focus on addressing these gaps to further elucidate the molecular mechanisms involved.
Conclusion
In conclusion, our research demonstrates that MG132 effectively suppresses cellular proliferation and induces apoptosis in ALL through the Akt/FOXO3a/Bim signaling pathway. By inhibiting Akt phosphorylation and reducing FOXO3a degradation, MG132 promotes the nuclear translocation of FOXO3a, leading to increased Bim expression (Figure 7). Additionally, MG132 significantly inhibited the growth of both primary cells and xenograft tumors. This study highlights MG132 as a promising therapeutic candidate for the treatment of ALL and provides a theoretical basis for its further exploration in clinical applications. MG132 activated the Akt/FOXO3a/Bim signal pathway by reducing the FOXO3a phosphorylation and decreasing the UPP degradation of FOXO3a.
Footnotes
Author contributions
This work was carried out in collaboration between all authors. L-N W, X-L Z and L-B H designed the experiments and revised the manuscript critically. Z F, W-H L, C L, Y L, C-J P, J-S L, W-Y T, L-M Z, D-P H, and Z-Y K designed methods and experiments, carried out most of the experiments. L-N W, X-L Z and L-B H analyzed data. Z F, W-H L, C L, Y L, C-J P, and J-S L wrote the manuscript. All authors approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (Grant No. 81600111 and 81600112), the Natural Science Foundation of Guangdong Province, China (Grant No. 2017A030313456), the Science and Technology Planning Project of Guangdong Province, China (Grant No. 2017A020215186 and 2016A020215045), the Medical Science and Technology Foundation of Guangdong Province, China (Grant No. A2019400 and A2019375), the Science and Technology Planning Project of Guangzhou, China (Grant No. 201707010004, 201604020128 and 201607010326).