
Abstract
Aristolochic acid (AA) is a compound extracted from the Aristolochia species of herbs. AA exposure is associated with kidney injury known as aristolochic acid nephropathy (AAN). Proximal tubular epithelial cell (PTEC) is the primary target of AA and rich in mitochondria. Recently, increasing evidence suggests that mitochondrial dysfunction plays a critical role in the pathogenesis of kidney disease. However, the status of mitochondrial function in PTEC after exposure to AA remains largely unknown. The aim of this study was to explore the effect of aristolochic acid I (AAI) on cell apoptosis and mitochondrial function in PTEC. Normal rat kidney-52E (NRK-52E) cells were exposed to different concentrations of AAI for different time periods. Cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide, cell apoptosis was analyzed by flow cytometry, and the expression of cleaved caspase-3 by Western blotting. Mitochondrial function was evaluated by reactive oxygen species (ROS), mitochondrial membrane potential (MMP), mitochondrial DNA (mtDNA) copy number, and adenosine triphosphate (ATP). It was found that AAI reduced cell viability and increased cell apoptosis in a dose- and time-dependent manner. In parallel to increased apoptosis, NRK-52E cell manifested signs of mitochondrial dysfunction in response to AAI treatment. The data indicated that AAI could increase ROS level, lower MMP, decrease mtDNA copy number, and reduce ATP production. In addition, Szeto-Schiller 31, a mitochondria-targeted antioxidant peptide, attenuated AAI-induced mitochondrial dysfunction and apoptosis. Our study depicted significant aberrant of mitochondrial function in AAI-treated NRK-52E cell, which suggested that mitochondrial dysfunction may be involved in AAI-induced apoptosis in PTEC.
Keywords
Introduction
Aristolochic acids (AAs) are a family of compounds derived from the genus Aristolochia, from the plant family Aristolochiaceae, which have been used for centuries to treat a variety of illnesses. 1,2 AA exposure will lead to kidney injury, which is known as aristolochic acid nephropathy (AAN). 3 AAN is characterized by rapidly progressive interstitial nephritis leading to end-stage renal disease and urothelial malignancy. 4 Although the sale and distribution of AA-containing supplements has been prohibited in most of the countries, AAN cases are still reported regularly all over the world. 5 Due to the selective uptake of AA via organic anion transporters (OATs), 6 proximal tubular epithelial cells (PTECs) are the preferential targets for the nephrotoxicity of AA. 7 The mechanisms of AAN include AA-DNA adducts formation, oxidative stress, apoptosis, inflammation, hemodynamic abnormalities, and fibrosis. 5 There is convincing evidence suggesting that cell apoptosis plays an important role in AAN pathogenesis. 8 –10 However, the potential mechanism of AA-induced apoptosis in PTECs still needs investigation.
Mitochondria are essential organelles for many aspects of cellular homeostasis, and mitochondrial dysfunction has been reported to be associated with metabolic, cardiovascular, and neurodegenerative diseases. 11 The reabsorption function of the proximal tubules makes it require a large amount of mitochondria to provide energy. 12 As such, they are more vulnerable to mitochondrial dysfunction from various insults. Swollen mitochondria in PTECs have been found in AAN patients, 13 acute AAN mice model, 10 and chronic AAN rat model. 14 Mitochondrial DNA (mtDNA) damage and mitochondrial permeability transition have also been reported to be associated with AA-induced nephrotoxicity. 15 –17 Moreover, colloidal gold immunoelectron microscopy revealed that aristolochic acid I (AAI) tended to accumulate in the mitochondria in renal tubules after treating BALB/c mice with AAI for 5 days. 18 These studies provided a clue for the role of mitochondrial dysfunction in AAN pathogenesis. However, the status of mitochondrial function in PTECs after exposure to AA has not been fully investigated. In the present study, Normal rat kidney-52E (NRK-52E) cells were exposed to different concentrations of AAI for different time periods. Cell viability, cell apoptosis, and markers of mitochondrial dysfunction were evaluated.
Materials and methods
Materials
AAI was purchased from Sigma-Aldrich (St Louis, Missouri, USA). Szeto-Schiller 31 (SS-31) was obtained from MedChemExpress (Monmouth Junction, New Jersey, USA). Dulbecco’s modified Eagle medium/nutrient mixture F12 (DMEM/F12) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, New York, USA). 3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was obtained from Invitrogen (Carlsbad, California, USA). The primary antibodies included rabbit cleaved caspase-3 (Asp 175) monoclonal antibody (Cell Signaling Technology, Beverly, Massachusetts, USA), mouse β-actin monoclonal antibody (Sigma-Aldrich), and mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (Proteintech, Wuhan, China). Horseradish peroxidase (HRP)-conjugated antimouse IgG and HRP-conjugated antirabbit IgG were purchased from Cell Signaling Technology (Beverly, Massachusetts, USA).
Cell culture and AAI treatment
NRK-52E cells were obtained from the American Type Culture Collection (Manassas, Virginia, USA). Cells were cultured in DMEM/F12 medium containing 10% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37°C with 5% CO2 in a humidified incubator. The cells were seeded in 35 mm dishes and grown to 80% confluence for subsequent experiments. After incubation with serum-free medium for 24 h, NRK-52E cells were exposed to different concentrations of AAI (0, 5, 10, 25, and 50 μM) for a specific time period or 50 μM AAI for different time periods (0, 4, 8, 12, and 24 h), respectively. At the end of experiments, the cells were used for analyses of cell viability, apoptosis, and mitochondrial function. To confirm the role of mitochondrial dysfunction in AAI-induced apoptosis, NRK-52E cells were pretreated with 1 μM SS-31, 19 a mitochondrial-targeted antioxidant peptide, in serum-free DMEM/F12 at 37°C for 1 h and then were exposed to 5 μM AAI for 24 h.
MTT assay
Cell viability was evaluated by MTT assay developed by Mosmann. 20 Cells were seeded at 105 cells/mL in 96-well plates and cultured with or without AAI. After AAI treatment, the medium was removed and replaced with 100-μL fresh culture medium. Then, 10-μL 12 mM MTT stock solution was added to each well and incubated the microplate at 37°C for 4 h. After labeling the cells with MTT, remove all but 25 µL of the medium from the wells, add 50 µL of dimethyl sulfoxide to each well and mix thoroughly, and incubate at 37°C for 10 min. The absorbance was measured at 570 nm using a microplate reader (BioTek, Winooski, Vermont, USA).
Annexin V fluorescein isothiocyanate/PI double staining
NRK-52E cells were seeded at 105 cells/mL in 35 mm dishes and treated with or without AAI. The cells were harvested after the incubation period and washed in cold phosphate-buffered saline (PBS). Then, cells were incubated with fluorescein isothiocyanate Annexin V and propidium iodide solution and analyzed the stained cells by flow cytometry following the manufacturer’s instructions (Invitrogen).
Western blotting
NRK-52E cells were seeded at 105 cells/mL in 35 mm dishes and treated with or without AAI. Cells were scraped and lysed in 100 μL of cellular lysis buffer (150 mM NaCl, 10 mM Tris–HCl, 5 mM ethylene diamine tetraacetic acid (EDTA), 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), and 10% Triton X-100) containing a protease inhibitor cocktail. Protein concentration was measured by the Bradford method and equal amounts of total protein from lysates were separated by 15% sodium dodecyl sulfate (SDS)-polyacrylamide gels and then transferred to polyvinylidene difluoride membranes (Millipore, Billerica, Massachusetts, USA). Membranes were blocked in 5% skimmed milk for 1 h at room temperature and then were incubated with rabbit anticleaved caspase-3 (1:200 dilution), mouse anti-GAPDH (1:5000 dilution), and mouse anti-β-actin (1:5000 dilution) antibody at 4°C overnight, respectively. The membranes were washed and incubated with appropriate HRP-conjugated secondary antibodies (1:2000 dilution) for 1 h at room temperature. The blots were visualized using an enhanced chemiluminescence system (Kodak Medical X-Ray Processor, Rochester, New York, USA). Densitometric analyses were performed using FluorChem8900 image analysis software (Alpha Innotech Corp., San Leandro, California, USA).
Reactive oxygen species production
For measurement of reactive oxygen species (ROS) production, NRK-52E cells (105 cells/mL) were stained with 2′,7′-dichlorofluorescein (DCF) diacetate, which is a cell-permeable nonfluorescent probe and turns to highly fluorescent form upon oxidation. The fluorescence intensity was collected using the flow cytometer. Data were expressed as the fold-increase of DCF fluorescence over control cells.
Mitochondrial membrane potential
The mitochondrial membrane potential (MMP) of NRK-52E cells was monitored using a lipophilic cationic probe, 5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) (Invitrogen). For quantitative analysis of MMP, JC-1 green and red fluorescence in NRK-52E cells (105 cells/mL) were recorded by flow cytometry at 525 and 600 nm, respectively. Mitochondrial depolarization was indicated by a decrease in the red/green fluorescence intensity ratio. 21
Quantitative real-time polymerase chain reaction
NRK-52E cells were seeded at 105 cells/mL in 35 mm dishes and treated with or without AAI. Total DNA from cultured NRK-52E cells was extracted by QIAamp DNA Mini Kit (QIAGEN, Germantown, Maryland, USA) following the manufacturer’s protocol. The concentration of extracted DNA was determined by spectrophotometry. For the estimation of mtDNA copy number, mitochondrial-encoded D-loop was amplified. The nuclear-encoded 18 S ribosomal ribonucleic acid (18 S rRNA) was used for normalization. 14 Primer sequences were as follows: D-loop: forward, 50-TGGTTCATCGTCCATACGTT-30; reverse, 50-TGACGGCTATGTTGAGGAAG-30 and 18 S rRNA: forward, 50-CATTCGAACGTCTGCCCTATC-30; reverse, 50-CCTGCTGCCTTCCTTGGA-30. Real-time polymerase chain reaction (PCR) amplification was performed using an ABI Prism 7900HTSequence Detection System (Life Technologies, Carlsbad, California, USA) with FastStart Universal SYBR Green Master (Roche, Basel, Switzerland). Cycling conditions were 95°C for 10 min followed by 40 repeats of 95°C for 15 s, 60°C for 30 s, and 72°C for 1 min. Fold changes of mtDNA copy number were calculated using the 2(− ΔΔCt) method.
Adenosine triphosphate content
NRK-52E cells were seeded at 105 cells/mL in 35 mm dishes and treated with or without AAI. Adenosine triphosphate (ATP) levels in NRK-52E cells were measured using ATP Colorimetric/Fluorometric Assay Kit according to the manufacturer’s protocol (Sigma-Aldrich). ATP content was determined by phosphorylating glycerol, resulting in a colorimetric (570 nm) or fluorometric (587 nm) product proportional to the amount of ATP present. Data were expressed as fold-changes over control cells.
Statistical analysis
All data were expressed as mean ± standard error of the mean from three separate experiments. Statistical differences were determined by two-tailed unpaired Student’s t-test or one-way analysis of variance (ANOVA) followed by post hoc analysis with Tukey’s test using SPSS statistics software version 16.0 (SPSS, Inc., Chicago, Illinois, USA). The value of p < 0.05 was considered as statistically significant.
Results
Effect of AAI on cell viability
MTT assay indicated that NRK-52E cell viability slightly increased after exposure to 5 μM AAI for 24 h (p < 0.05). As the concentration of AAI increased, cell viability gradually decreased and reached statistical significance at 50 μM AAI (p < 0.05). Then, we treated NRK-52E cells with 50 μM AAI for different time periods. The data showed that cell viability decreased significantly after 4 h of stimulation and gradually decreased over time (Figure 1).

(a) Chemical structure of AAI, (b) effect of AAI on NRK-52E cell viability at the dose point, and (c) time point. Data are presented as (percentage of control) mean ± SEM of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control cells. AAI: aristolochic acid I; SEM: standard error of the mean.
Effect of AAI on cell apoptosis
Flow cytometry found that 24 h AAI exposure increased the proportion of apoptotic cells in a dose-dependent manner. However, 50 μM AAI stimulation did not markedly increase the proportion of apoptotic cells at different time periods until the stimulation time was extended to 24 h (33.26 ± 0.44% vs. 4.78 ± 0.73%, p < 0.001; Figure 2). The protein expression of cleaved caspase-3, a marker of cell apoptosis, was further detected by Western blotting. As shown in Figure 3, cleaved caspase-3 was significantly upregulated under all set AAI stimulation conditions.

Effect of AAI on apoptosis in NRK-52E cell. (a) Flow cytometry scatter diagram obtained via Annexin V FITC and PI double staining. (b and c) Column bar graphs of the apoptotic rate of each group. Data are presented as mean ± SEM of three separate experiments. **p < 0.01, ***p < 0.001 compared with the control cells. FITC: fluorescein isothiocyanate; PI: propidium iodide; AAI: aristolochic acid I; SEM: standard error of the mean.

Effect of AAI on the expression of cleaved caspase-3. (a) Representative Western blot images of proapoptotic protein cleaved caspase-3. (b and c) Densitometric analysis of cleaved caspase-3 protein expression normalized to GAPDH content. Data are presented as mean ± SEM of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control cells. GAPDH: glyceraldehyde-3-phosphate dehydrogenase; AAI: aristolochic acid I; SEM: standard error of the mean.
Effect of AAI on mitochondrial function
Mitochondrial function was evaluated by ROS, MMP, mtDNA copy number, and ATP content. Our data showed that the level of intracellular ROS reached its highest value after 15 min of stimulation with 50 μM AAI. Using 15 min as the longest stimulation time, ROS levels gradually increased with increasing AAI concentration (Figure 4). After exposure to AAI, MMP in NRK-52E cells decreased significantly in a dose- and time-dependent manner (Figure 5). mtDNA copy number decreased only significantly after 50 μM AAI stimulation for 24 h. However, stimulation of NRK-52E cell with 50 μM AAI significantly reduced mtDNA copy number at different time periods (Figure 6). As shown in Figure 7, AAI treatment for 24 h obviously lowered ATP content in NRK-52E cell at any concentrations. However, stimulation of NRK-52E cell with 50 μM AAI for 4, 8, or 12 h had little effect on intracellular ATP content (Figure 7).

Effect of AAI on the level of ROS in NRK-52E cell. (a) Flow cytometry diagram obtained via DCFDA staining. (b and c) Quantitative analysis of DCF fluorescence of each group. Data are presented as mean ± SEM of three separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control cells. DCFDA: 2′,7′-dichlorofluorescin diacetate; DCF: 2′,7′-dichlorofluorescein; AAI: aristolochic acid I; SEM: standard error of the mean; ROS: reactive oxygen species.
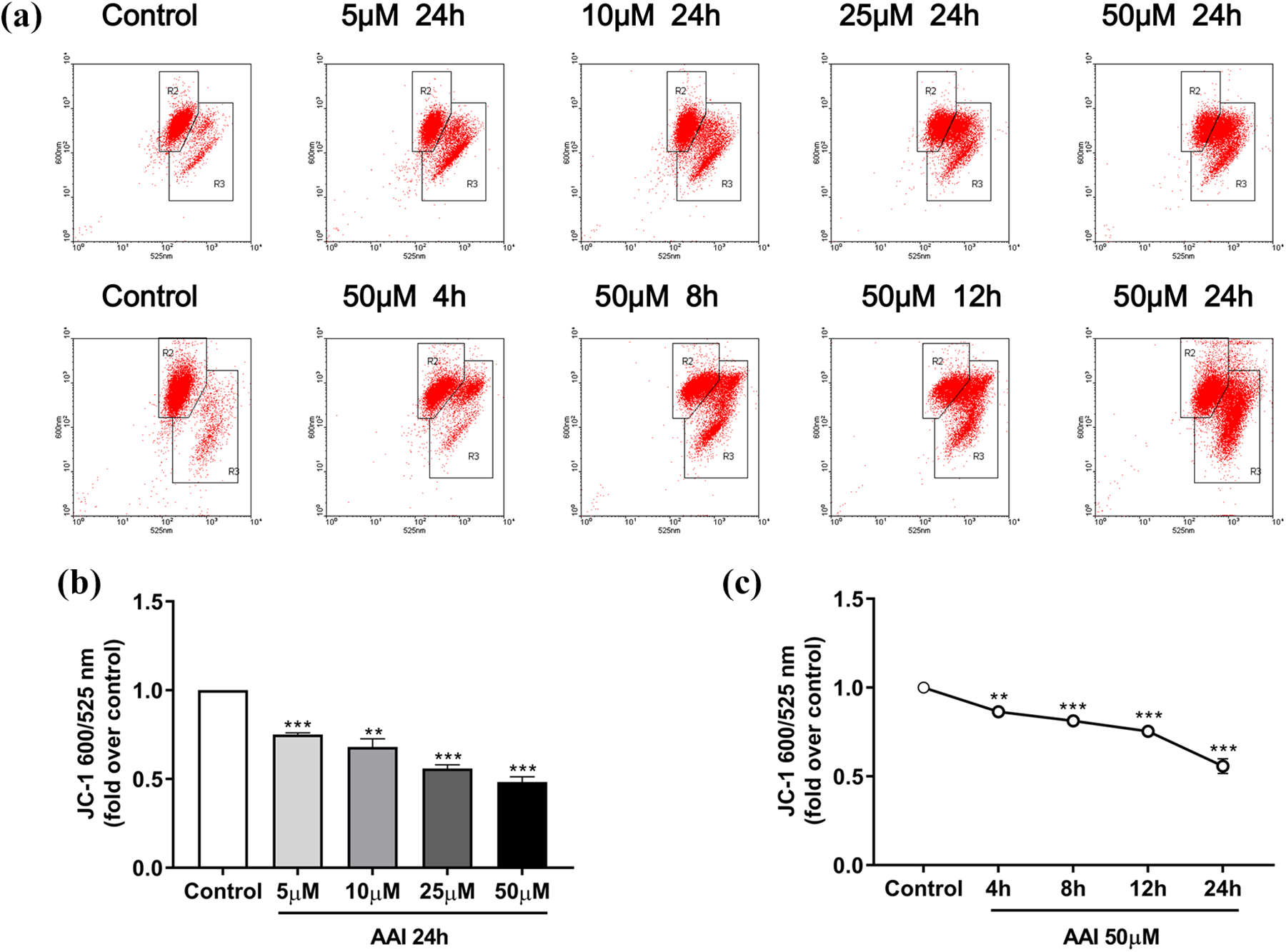
Effect of AAI on MMP in NRK-52E cell. (a) Flow cytometry diagram obtained via JC-1 staining. (b and c) Quantitative analysis of JC-1 600/525 nm of each group. Data are presented as mean ± SEM of three separate experiments. **p < 0.01, ***p < 0.001 compared with the control cells. JC-1: 5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; AAI: aristolochic acid I; SEM: standard error of the mean; MMP: mitochondrial membrane potential.

Effect of AAI on mtDNA copy number in NRK-52E cell. (a) mtDNA copy number after exposure to different doses of AAI for 24 h and (b) mtDNA copy number after exposure to 50 μM AAI for different time periods. Data are presented as mean ± SEM of three separate experiments. **p < 0.01, ***p < 0.001 compared with the control cells. mtDNA: mitochondrial DNA; AAI: aristolochic acid I; SEM: standard error of the mean.

Effect of AAI on ATP content in NRK-52E cell. (a) ATP content after exposure to different doses of AAI for 24 h and (b) ATP content after exposure to 50 μM AAI for different time periods. Data are presented as mean ± SEM of three separate experiments. ***p < 0.001 compared with the control cells. ATP: adenosine triphosphate; AAI: aristolochic acid I; SEM: standard error of the mean.
Effect of SS-31 on AAI-induced mitochondrial dysfunction and apoptosis
As shown in Figure 8, pretreatment with SS-31 markedly inhibited AAI-induced ROS production and protected against ATP reduction after exposure to AAI. Moreover, the protein expression of cleaved caspase-3 was significantly suppressed in the AAI + SS-31 group (Figure 8(c) and (d)).

SS-31 protected against AAI-induced mitochondrial dysfunction and apoptosis in NRK-52E cell. (a) The level of reactive oxygen species was determined by quantitative analysis of DCF fluorescence, (b) ATP content in each group. (c) representative Western blot images of cleaved caspase-3, and (d) densitometric analysis of cleaved caspase-3 protein expression normalized to β-actin content. Data are presented as mean ± SEM of three separate experiments. ***p < 0.001 compared with the control group; #p < 0.05, ###p < 0.001 compared with the AAI group. ATP: adenosine triphosphate, DCF: 2′,7′-dichlorofluorescein, SS-31: Szeto-Schiller 31; AAI: aristolochic acid I; SEM: standard error of the mean.
Discussion
In the present study, NRK-52E cells were exposed to AAI at different concentrations for different time periods. Cell viability, cell apoptosis, and mitochondrial function were evaluated after AAI treatment. The results suggested that AAI could reduce cell viability, induce cell apoptosis, and impair mitochondrial function in NRK-52E cell.
AAN is marked by elevated serum creatinine, significant anemia, and rapid progression to end-stage renal disease. In pathology, predominantly proximal tubular cells demonstrate loss of brush border and atrophy progressing from the superficial to the deep cortical regions. 22 There is compelling evidence demonstrating that apoptosis is involved in tubular atrophy, renal fibrosis, and the progression to chronic kidney disease. 5 Previous studies have reported that AAI could induce apoptosis in cultured tubular epithelial cells from different species, including LLC-PK1 (pig), 23 mProx24 (murine), 24 and Madin–Darby canine. 25 Similar to the research of Wu et al., 26 our study confirmed the role of AAI in inducing apoptosis by using rat kidney tubular epithelial cell line NRK-52E. These studies collectively highlighted the important role of apoptosis in AA-induced nephrotoxicity.
The kidney has the second highest mitochondrial content and oxygen consumption after the heart. 12 Proximal tubules require a large amount of mitochondria due to their high-energy demand for reabsorption and secretion against chemical gradients. 27 As such, they are more vulnerable to suffer from mitochondrial dysfunction. 28,29 Since PTEC is the primary target for the nephrotoxicity of AA, it is reasonable to speculate that mitochondrial dysfunction is involved in AAI-induced apoptosis in PTEC. In our study, four key indicators were used to evaluate AA-induced mitochondrial dysfunction in NRK-52E cells. The results indicated that exposure to AAI could increase ROS level, lower MMP, decrease mtDNA copy number, and reduce ATP production. Mitochondrial respiratory chain is the primary source of intracellular ROS. 30 Overproduction of ROS will damage respiratory chain and mtDNA, ultimately leading to mitochondrial dysfunction. 31 Previous studies have reported that ROS was associated with AAI-induced oxidative DNA damage, 32 apoptosis, 26 and cell cycle arrest. 33 The MMP generated by proton pumps is an essential component in the process of driving force for ATP synthesis in mitochondria. 34 Both MMP and ATP production were marked impaired in NRK-52E cells in our study. Mitochondrion is the only organism that has inheritable DNA, which is called mtDNA. mtDNA encodes several mitochondrial respiratory protein expression, which is indispensable for mitochondrial function. mtDNA lacks protection by histone proteins and efficient repair processes. 35 Damage to mtDNA will affect the expression of its encoded respiratory chain proteins, leading to mitochondrial dysfunction. In line with previous studies in acute and chronic AAN rat model, 14,15 our results also showed a significant decrease of mtDNA copy in NRK-52E cell exposure to AAI. In addition, pretreatment with SS-31, a mitochondria-target antioxidant peptide, inhibited AAI-induced ROS production, ATP reduction, and cleaved caspase-3 expression, which validated the role of mitochondrial dysfunction in AAI-induced apoptosis in NRK-52E cell. Previously, SS-31 has been reported to be beneficial for the treatment of acute kidney injury, 36 diabetic nephropathy, 37 and renal fibrosis. 38 The mitoprotective effect of SS-31 in the treatment of AAN needs further investigation.
In summary, our data suggest that mitochondrial dysfunction is involved in AAI-induced apoptosis in NRK-52E cells. These results provide new insight into the possible mechanism and potential therapeutic target for AAN.
Footnotes
Author contribution
XL and JW contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of China (Grant Nos. 81774069, 81570614, 81603437, 81600516, and 81973602), National Key Research and Development Program (Grant No. 2016YFC0906101), the Guangdong Natural Science Foundation of China (Grant Nos. 2014A030313139 and S2011010005077), Foundation of Guangdong Key Laboratory of Nephrology (Grant Nos. 2014B030301023 and 2017B030314019), and the Guangzhou Committee of Science and Technology, China (Grant Nos. 2014Y2-00543 and 201704020167). The funders of this study had no role in study design; collection, analysis, and interpretation of data; writing the report; and the decision to submit the report for publication.