
Abstract
Doxorubicin (DOX) is an antineoplastic agent obtained from Streptomyces peucetius. It is utilized in treating different kinds of cancers, such as leukemia, lymphoma, and lung, and breast cancers. The main side effect of DOX is cardiotoxicity. Metformin (MET) is an antihyperglycemic drug used for type 2 diabetes treatment. It is proposed that MET has a protective effect against DOX cardiotoxicity. Our review demonstrated that MET has several possible mechanisms of action, which can prevent or at least reduce DOX cardiotoxicity including a decrease of free radical generation and oxidative stress, 5′ adenosine monophosphate-activated protein kinase activation, and ferritin heavy chain expression in cardiomyocytes cells. The combination of MET and DOX has been shown to enhance the anticancer activity of DOX by a number of authors. The literature reviewed in the present report supports the hypothesis that MET can reduce the cardiotoxicity that often occurs with DOX treatment.
Introduction
Doxorubicin (DOX) (Adriamycin) is an anthracycline chemotherapeutic drug that is used alone or with other treatments/medications to treat several different types of cancer, including breast cancer, bladder cancer, Kaposi’s sarcoma, Hodgkin’s and non-Hodgkin’s lymphoma, and certain types of lung cancer. 1 DOX is given by injection into a vein. Common side effects of DOX are included hair loss, bone marrow suppression, vomiting, rash, and inflammation of the mouth. More serious side effects may include allergic reactions such as anaphylaxis, heart damage (cardiomyopathy), tissue damage at the site of injection, radiation recall, and treatment-related leukemia. People often experience red discoloration of the urine for a few days following the administration of drug. 2,3
Although the exact mechanism of DOX remains under investigation, it is believed that DOX interacts with DNA, disturbing macromolecular biosynthesis. 4 –6 It has been suggested that DOX induces cardiomyopathy by increasing oxidant production in the heart. 7 –11 The mitochondria appear to be the primary target of DOX cardiotoxicity, resulting from single electrons being transmitted to DOX with the formation of oxygen radicals through autoxidation processes. Evidence further suggests that a nicotinamide adenine dinucleotide phosphate (NAD(P)H) dehydrogenase is associated with the complex I electron transport chain involved in the electron transfer to DOX. 12,13
Some of the cardiotoxic signs induced by DOX include hypotension, tachycardia, transient arrhythmias, and late-onset cardiomyopathy in the form of fatigue, dyspnea, and lower limb edema. 14
Metformin (MET) is a biguanide, a family of compounds that share a similar chemical form, that is widely used for type 2 diabetes. 15 Its antihyperglycemic effect is not related to the stimulation of insulin secretion but its effect on peripheral tissues makes these tissues more sensitive to the action of insulin. In vitro and in vivo studies have shown that the effect of MET on glucose level is mainly due to reducing hepatic glucose output and increasing peripheral glucose uptake. 16 –21 The peripheral mechanism of MET depends on the glucose transporter system. 22 A recent epidemiological study in diabetic cancer patients has suggested that MET users have a lower risk for cancer in comparison to patients using insulin or an insulin stimulator. Studies in animal tumor models and cancer cell lines have shown that MET prevents tumor development. 23
Antioxidant activity of MET in cardiomyocytes is accomplished via adenosine monophosphate-activated protein kinase (AMPK) activation, which reduces the production of reactive oxygen species (ROS) in animal models of heart failure. 24,25 MET may protect cardiomyocytes from oxidative stress induced by H2O2 or tumor necrosis factor-alpha (TNF-α). 26 Recently, MET has been shown to prevent oxidative stress and DOX-induced cell death in cardiomyocytes. 27
Due to the extensive use and therapeutic potential of MET in humans and especially in diabetic patients, this article reviews the potential cardioprotective mechanisms of MET in individuals treated with DOX as an antineoplastic drug (Table 1).
Summary of protective activities of MET against DOX-induced cardiotoxicity.

i.p.: intraperitoneal; MDA: malondialdehyde; DOX, doxorubicin; MET: metformin; LDH: lactate dehydrogenase; CK-MB: creatine kinase-muscle/brain; COX: cyclooxygenase; SOD: superoxide dismutase; TBARS: thiobarbituric acid reactive substance; PDGFR, platelet-derived growth factor receptor; iNOS: inducible nitric oxide synthase; TNF: tumor necrosis factor; ROS: reactive oxygen species; FHC: ferritin heavy chain; mRNA: messenger RNA; ATP: adenosine triphosphate; TGF-ß, transforming growth factor-ß; LV: left ventricular; AMP: 5′ adenosine monophosphate-activated protein; EF: ejection fraction; CAT: catalase; GPx: Glutathione peroxidase; CoA, CoASH: Coenzyme A.
Literature search
We reviewed the following databases PubMed, Scopus, ScienceDirect, and Web of Science, using the following search terms (keywords) doxorubicin, cardiotoxicity, metformin, Adriamycin, and heart failure either in the title, the abstracts, or in the text over the years 1980 to February 2019. Only articles in English were reviewed and the relevant publications were further evaluated based on title and abstract. The final version of this peer review was based on the selected articles.
DOX toxicity mechanisms and protective effects of MET
Some of the more important molecular mechanisms reported for DOX cardiotoxicity are oxidative stress, mitochondrial dysfunction, and dysregulation of autophagy. 28
Oxidative stress
Much of the literature suggests that the main mechanism of DOX cardiotoxicity is oxidative stress 29 –31 which can be transmitted through various routes including mitochondrial, nitric oxide synthase, and NAD(P)H. The mitochondria are reported to be the main target of cationic drugs like DOX at the subcellular level because DOX forms an irreversible complex with cardiolipin on the inner membrane of the mitochondria. 32 The DOX cardiolipin complex produces superoxide (O2 −) anions and induces oxidative stress. 33 A significant reduction of long-chain fatty acid oxidation in cardiac mitochondria while glucose metabolism was increased following the infarct with DOX was shown in an in vitro study. This signaled a general shift from aerobic to anaerobic metabolism. Although it is a common feature of heart failure, the DOX driven shift from long-chain fatty acid metabolism to glucose metabolism occurs when these metabolic genes are transcriptionally suppressed. 34 This functional disruption can lead to ultrastructural pathologic changes such as mitochondrial swelling and myelin bodies within the mitochondria. 35,36 Another in vitro study showed that MET increased aerobic glycolysis and reduced glucose metabolism through the citric acid cycle potentially leading to attenuated DOX cardiotoxicity. 37
One in vitro experiment on HL-1 cells showed that MET administration increased the expression of adiopoR1 and adipoR2. 38 AdipoR1 stimulated the activity of AMPK and AdipoR2 increased cellular insulin sensitivity. As a result, AdipoR1 prevented cell death due to energy shortages and AdipoR2 inhibited the cellular metabolism switch from aerobic to anaerobic, thereby reducing free radicals generation. 39,40
DOX stimulates the apoptosis pathway through mechanisms that do not directly capture ROS and oxidative stress. In this pathway, oxidative stress activates heat shock factor 1 (HSF-1). This protein is the main intermediary of the transcriptional reaction to proteotoxic stress. 41 HSF-1 acts to produce more heat shock protein 25 (HSP25) and consequently results in the stabilization of p53. This gene is a classical tumor suppressor gene that activates apoptosis. 42 One study revealed that the knockdown of HSP25 decreased the interaction between sirtuin (silent mating type information regulation 2 homolog) 1 (Saccharomyces cerevisiae, SIRT1, an enzyme that reacts to stressors 43 ) and p53. This reaction led to an increased p53 acetylation on lysine 379 (K379) and consequently upregulated pro-apoptotic Bax protein expression. These mechanisms evaluated by H9c2 cells. 44 Also, it showed that overexpression of HSP25 arrested the cell cycle in murine L929 cells. 45 Expression of SIRT1 inhibited the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-regulated gene expression and increased insulin sensitivity. 46 –50 These investigators showed that MET can attenuate DOX side effects by SIRT1 expression through decreased inflammatory cytokines and increased insulin sensitivity. 51,52
One of the cardiotoxicity mechanisms of DOX is toll-like receptor (TLR) activation. In in vivo studies on the mouse, rat, rabbit, swine, and dog, the role of TLR2 and TLR-4 in the development of cardiomyopathy was evaluated. TLR-2-mediated signaling occurred through the pro-inflammatory NF-κB pathway which also involved cytokine production, apoptosis, and cardiac dysfunction. TLR-4 activation leads to the NF-κB intracellular signaling pathway producing inflammatory cytokines that are responsible for activating the inherent immune system. 53 MET inhibited interleukin (IL)-1β dose-dependently in human vascular wall cells. This cytokine induced release of pro-inflammatory cytokines such as IL-6 and IL-8 in endothelial cells, smooth muscle cells, and macrophages. The results showed that MET reduced IL-1β-induced activation and nuclear translocation of NF-κB. In addition, MET prevented IL-1β-induced activation of pro-inflammatory phosphokinases such as Akt, p38, Erk, and protein kinase C. 54
MET can prevent inflammatory stress by suppression of TLR-2 and TLR-4 in cardiac tissue. 55 –57 Moreover, it has been reported that MET mediated AMPK activation 58 and prevented apoptosis induced by DOX through TNF-α and caspase-3 inhibition. AMPK activation resulted in a rapid and sustained increase in BCL2 associated agonist of cell death (BAD) phosphorylation. This event reduced the interaction between BAD and B-cell lymphoma-extra large (Bcl-xL), limiting cytochrome c release and caspase-3 activation. 26,59,60 MET, during the reperfusion state, induced rapid activation of Akt. This activation inhibited the mitochondrial permeability transition pore (mPTP) opening. 61 Although the mechanism of the relation between Akt and mPTP is unclear, it could be related to the effect of Akt on cellular calcium handling. 62 or its downstream targets such as glycogen synthase kinase 3 beta. 63 mPTP is a nonspecific pore of the inner mitochondrial membrane, which is believed to open in the first few minutes of myocardial reperfusion in response to adenosine triphosphate (ATP) drainage and oxidative stress. 64,65 The opening of the mPTP incites cardiomyocyte death by uncoupling oxidative phosphorylation and mitochondrial swelling. 66 Therefore, MET by increasing Akt activity inhibited ATP starvation and mPTP opening and reduced DOX cardiotoxicity. 61
DOX administration increased the cardiac levels of malondialdehyde (MDA) and nitric oxide and induced a reduction in cardiac superoxide dismutase (SOD) activity. Coadministration of MET with DOX resulted in a significant decrease in serum levels of lactate dehydrogenase and creatine kinase-muscle/brain enzymes and cardiac MDA as well as total nitrites and nitrates levels. In addition, an increase in cardiac SOD activity was observed. 67,68
The results of another study on male Wistar albino rats demonstrated that the administration of DOX (20 mg/kg, intraperitoneal (i.p.)) for 7 days showed a significant reduction in cardiac glutathione (GSH), which is associated with increased cardiac thiobarbituric acid reactive substance (TBARS). The cardiac TNF-α level also was significantly elevated. Administration of MET (500 mg/kg/day, p.o.) to DOX-intoxicated rats resulted in an increase in GSH and a decrease in TBARS and TNF-α levels. 69
These MET effects were observed in myocardial function including aortic flow, cardiac output, and histopathological improvement. 68 Another study revealed that the combination of MET (250 mg/kg/day, p.o.) and DOX (4 mg/kg, twice a week, i.p.) for 14 days improved left ventricular end systolic, interventricular septum thickness, ejection fraction, and fractional shortening in Wistar albino rats. 70,71
FHC and DOX
The DOX-iron complex has been known since 1980 when studies reported that DOX had a strong affinity for iron. 72 The iron complex induces lipid peroxidation. 73 The reduction of DOX in the presence of free iron initiates a cycle of free radical production. Doxorubicinol (DOXol) is the DOX metabolite that interacts with the thiol group of proteins. 74 More recent studies have shown that the effect of DOX on iron metabolism is not affected by the DOX-iron interaction but by its effect on iron regulatory proteins (IRPs). One mechanism involves the DOXol metabolite forming a complex with the Fe–S group of the cytoplasmic aconitase/IRP-1. This complex increases the stability of transferrin messenger RNA (mRNA) and prevents the translation of the iron sequestration proteins. 75 The subsequent decrease in IRP-1 leads to an increase in free iron which maintains the free radical production cycle. Therefore, interference with iron sequestration is a critical component of DOX-induced cardiotoxicity. 76
Studies have indicated that an increase in the free iron pool by DOX was prevented by MET through upregulation of the ferritin heavy chain (FHC) and consequently iron chelation. 77,78 FHC is the main iron-storage protein. 79 MET upregulates FHC via NF-κB activation in cardiac cells. Also, MET prevented sustained c-Jun N-terminal kinase (JNK) activation by FHC mediation and, thereby, apoptosis was induced by TNF-α. 80 DOX (5 μM) administration in HL-1 cells stimulated cytochrome C release from mitochondria into the cytosol and MET (4 mM) prohibited this effect by FHC mediation. Another in vitro study with the same protocol confirmed that MET prevented DOX-induced oxidative stress through FHC activation and ROS suppression. 77,78
Intracellular calcium dysregulation
Another reported mechanism of DOX cardiotoxicity is increasing the intracellular calcium level. Dysregulation of the intracellular calcium concentration causes ROS production. 81 It has been shown that DOX-induced calcium liberation from the sarcoplasmic reticulum occurs. 82 It has been suggested that DOX has a high affinity for the ryanodine receptor and that DOX blocked the sodium–calcium exchanger in the sarcolemma 83 and increased the L-type calcium channel activity. 84 These findings suggested that calcium dysregulation is important in the pathogenesis of DOX-induced cardiomyopathy.
A study has reported that coadministration of DOX (10 nM) with MET at low concentration (0.1 mM) reduced the [Ca2+]i by decreasing ROS generation in H9c2 cells. But at higher concentrations (1.0 mM), MET lacks sufficient protective effect. These findings suggested a dual effect of MET in the cardioprotective process. This effect is apparently dependent on platelet-derived growth factor receptor (PDGFR) expression level. It has been reported that PDGFR signaling promotes hypoxia-induced macroautophagy in tumor cells. At lower concentrations, MET increased AMPK and consequently PDGFR activity but at higher concentrations, MET led to extra AMPK activity in the cells. At these higher doses, MET resulted in suppression of PDGFR and the protective effect of MET was reversed or at least reduced. 85
Changes in high-energy phosphate pool
Disturbance of mitochondrial energy production is an important adverse effect of DOX, which can lead to apoptosis and mitochondrial dysfunction. The mitochondrial damage may manifest itself as a decrease in ATP production. The lack of ATP reduces the affinity of HSP90 86 for erythroblastic oncogene B (ERBB2), a cardioprotective protein. 87 ERBB2 signaling is a response to the neuregulin ligand, which regulates anthracycline uptake into cells. Therefore, upregulation of cardiac neuregulin signaling may be one strategy to limit myocardial anthracycline damage. 88 When ATP is decreased, the ERBB2 level will be reduced and HSP90 can no longer maintain its chaperone role. Since ERBB2 is connected to various G protein-coupled receptors, this transactivation to the pro-survival ERK1/2 pathway is interrupted. 89,90 These findings suggested that the MET effect on cells was mediated by AMPK activation. One study on a human tumor xenograft model reported that MET induced a moderate decrease in ATP levels which resulted in AMPK activation. 91 AMPK activation stimulated muscle glucose uptake and reduced energy starvation. 92 In the case of ERBB2 overexpression, MET at higher concentrations reduced this expression, while at lower concentrations, MET inhibited ERBB 2 tyrosine kinase activity. 93 –95 MET also has been reported to increase the HSP90 level. 96
Extracellular matrix remodeling
Although the intracellular matrix has a very important role in cardiomyocytes reperfusion, changes in the structural features of the extracellular matrix also can affect cardiac tissue amendment. 97 DOX inhibited transcription and translation of the collagenase/matrix metalloproteinase 1 (MMP-1) in tumoral cells. MMP-1 breaks down types I, II, and III interstitial collagens. 98 However, DOX showed a different effect on heart tissue because it increased the production of MMP-2 and MMP-9. 99,100 It has been suggested that cardiac tissue was improved by reducing the MMPs level in the cardiomyocytes. MMPs selectively degrade various components of the extracellular matrix. The MMPs are also capable of inducing cytokines and chemokines. Therefore, MMPs appear to play important roles in inducing inflammation in cardiomyocites. Both MMP-2 and MMP-9 activities have been reported to be enhanced by ROS production. 101 MET caused a significant decrease in both the MMP-2 and MMP-9 mRNA levels in a concentration-dependent manner. 102 This suppression attenuated cardiac hypertrophy. 103
Autophagy dysregulation
Autophagy (or autophagocytosis) (from the Greek auto-, “self,” and phagein, “to eat”) is the natural, destructive mechanism that disassembles, through a regulated process, unnecessary or dysfunctional cellular components. It is a survival mechanism that destroys cellular components to maintain the health of damaged cells. 104,105 The three types of autophagy in mammalian cells are macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy proceeds by the generation of an initiation complex containing UNC-51 like kinase 1 (ULK1), FIP200, and the autophagy-related gene (Atg) 13 to form the preautophagosomal double-lipid bilayer. Specifically, ULK1-mediated phosphorylation of Beclin-1 at serine 14 enables activation of the vacuole sorting protein (Vps) 34 and its coactivator Vps15. Vps34–Vps15 activation induces phosphoinositol triphosphate accumulation that, in turn, promotes the WD-repeat interacting with phosphoinositides protein leading to the developing autophagosomal complex in the inner membrane. 106
The role of autophagy in DOX-induced cardiotoxicity has recently been explored. However, there are conflicting reports but most of the reports have suggested that DOX upregulates cardiac autophagy. DOX induces autophagy by suppressing the expression of GATA Binding Protein 4 and/or S6K1, which stimulates the expression of autophagy genes such as Atg12, Atg5, Beclin1, and Bcl-2. 107 It was observed that the administration of MET resulted in an increase of the Beclin-1 level. 28 Beclin-1 is a mammalian protein that is involved in both autophagy and cell death regulation. 108 Administration of MET (250 mg/kg/day, p.o.) with DOX (3 mg/kg, every second day, i.p.) for 2 weeks inhibited the elevation of this protein. The decrease in light chain 3B (LC3-II) was observed in the DOX-treated group. LC3 is a central protein in the autophagy pathway that plays a role in autophagy substrate selection and autophagosome biogenesis. 109 Also, coadministration of MET with DOX resulted in LC3 expression. An elevated level of p62 (a protein that recognizes toxic cellular waste 110 ) in the DOX-treated group was alleviated by MET. 28
The various mechanisms discussed above are summarized in Figure 1.
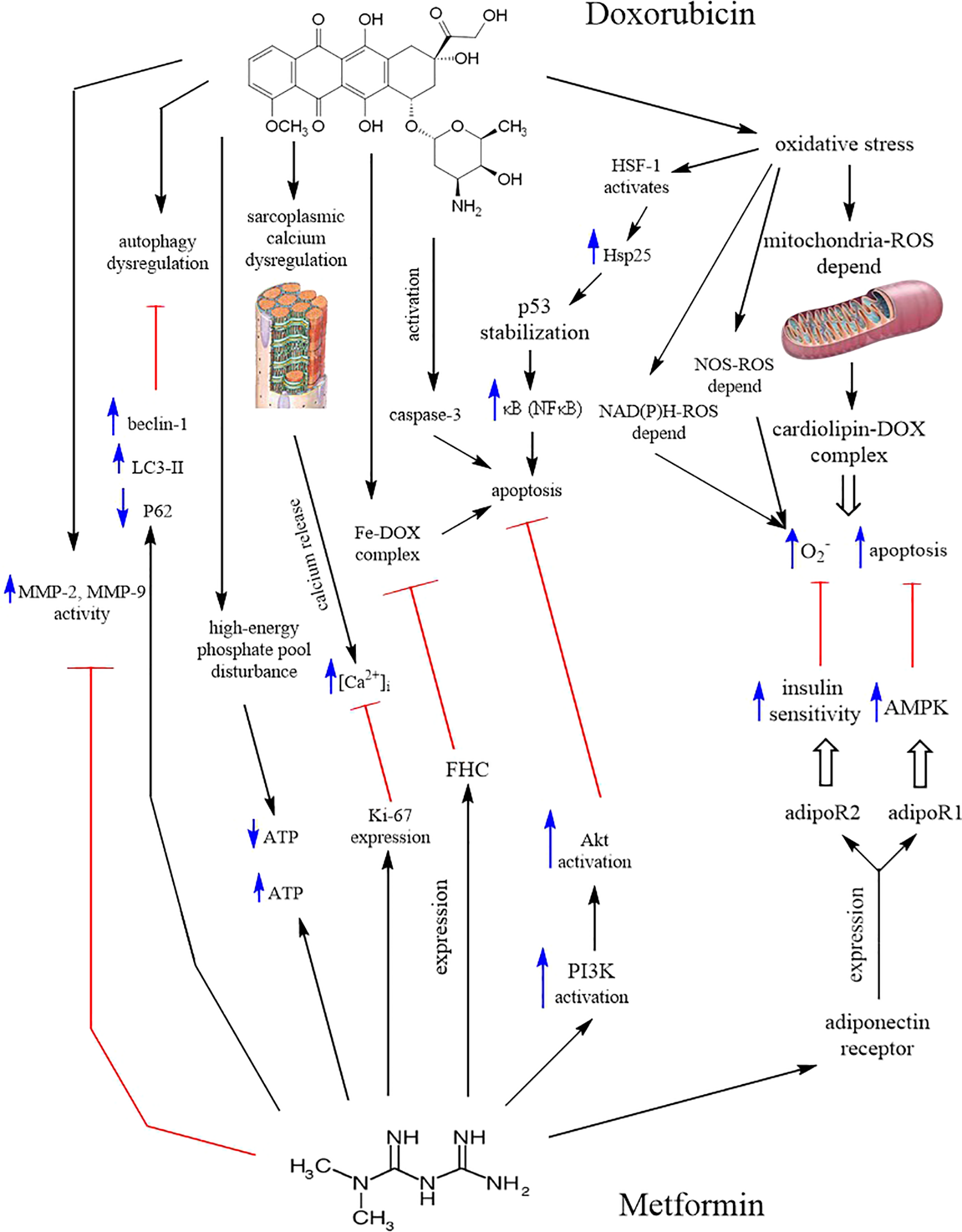
Summary of DOX cardiotoxic mechanisms and MET intervention. DOX: doxorubicin; MET: metformin; LC3: light chain 3B; ROS: reactive oxygen species; NOS: nitric oxide synthase; NAD(P)H: nicotinamide adenine dinucleotide phosphate; FHC: ferritin heavy chain; MMP: matrix metalloproteinases; NF-κB: nuclear factor kappa B; HSF-1: heat shock factor 1; Hsp25: heat shock protein 25; PI3 K: phosphatidylinositol-3 kinase; AMPK: 5′ adenosine monophosphate-activated protein kinase; ATP: adenosine triphosphate.
Conclusion
The current report reviewed the available peer-reviewed literature that investigated the various cardiotoxicity mechanisms of DOX and the potential protective effects of MET. Oxidative stress is one of the most important mechanisms of DOX-induced heart damage. This drug also has been shown to change aerobic metabolism to anaerobic metabolism. MET can increase the cellular energy level by activation of AMPK, thus increasing the rate of cell survival.
Iron hemostasis plays an important role in oxidative stress induced by DOX in the heart. Increasing the cytosolic and mitochondrial free iron pools induced by DOX can be prevented or at least reduced by MET through FHC expression. DOX has been shown to liberate calcium from the sarcoplasmic reticulum and MET can reduce the [Ca2+]i and thereby attenuated the DOX cardiotoxicity. DOX can induce autophagy dysregulation which has been reported to be alleviated by the use of MET.
It is an important scientific challenge to find medication when combined with DOX could decrease cardiotoxicity, while maintaining the efficacy and safety of DOX in cancer therapy.
MET may be a promising approach for patients treating with DOX.
Footnotes
Acknowledgment
The authors are thankful to Mashhad University of Medical Sciences.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.