
Abstract
Background:
Nicotine and cigarette smoking (CS) are associated with addiction behavior, drug-seeking, and abuse. However, the mechanisms that mediate this association especially, the role of brain-derived neurotrophic factor (BDNF), dopamine (DA), and nuclear factor erythroid 2-related factor 2 (Nrf2) signaling in the cerebral cortex, are not fully known. Therefore, we hypothesized that overexpression of BDNF and DA, and suppression of Nrf2 contribute to several pathological and behavioral alterations in adult cerebral cortex.
Methodology/Principal Observations:
We treated Wistar rats with different doses of oral nicotine and passive CS for 4-week (short-term) and 12-week (long-term) duration, where doses closely mimic the human smoking scenario. Our result showed dose-dependent association of anxiogenic and depressive behavior, and cognitive interference with neurodegeneration and DNA damage in the cerebral cortex upon exposure to nicotine/CS as compared to the control. Further, the results are linked to upregulation of oxidative stress, overexpression of BDNF, DA, and DA marker, tyrosine hydroxylase (TH), with concomitant downregulation of ascorbate and Nrf2 expression in the exposed cerebral cortex when compared with the control.
Conclusion/Significance:
Overall, our data strongly suggest that the intervention of DA and BDNF, and depletion of antioxidants are important factors during nicotine/CS-induced cerebral cortex pathological changes leading to neurobehavioral impairments, which could underpin the novel therapeutic approaches targeted at tobacco smoking/nicotine’s neuropsychological disorders including cognition and drug addiction.
Keywords
Introduction
Drug addiction is a chronically relapsing disorder characterized by compulsive drug-seeking and abuse that affect neurocircuit involving cognitive and mood-associated behaviors, probably due to the pathological changes in different brain regions. 1 As per the USDHHS, cigarette smoking (CS) is a powerful addictive drug and nicotine is one of the major pharmacologically active components of CS, 2 which degenerates midbrain dopaminergic (DAergic) neurons by rapid activation of tyrosine hydroxylase (TH) and inhibition of monoamine oxidase-B. 3 Nicotine modulates DAergic system targeting dopamine D1 and D2 receptors (DA D1 and D2 Rs) in the caudate putamen (cPU) of the adult rat brain. 4 Acute doses of nicotine facilitate DA release, but chronic nicotine treatment decreases DA turnover in striatum. 5 Further, like DA, brain-derived neurotrophic factor (BDNF) is another major neurochemical, which influences serotonin and DAergic neurotransmission in the ventral tegmental area (VTA)-nucleus accumbens (NAcc) pathway that involves reward system of addiction. 6 Nicotine-induced release of BDNF and upregulation of its receptor (Trkβ) are also suggested in the rat brain. 7 Moreover, high synaptic DA induces BDNF expression in the adult rat midbrain to initiate addiction behavior such as, stress and depression via DA D1 R activation in drug/substance abuse cases. 8 Acute Trkβ activation has been found to potentiate presynaptic DA release and its transport in different brain regions. 9 Interestingly, the activation of Trkβ releases intracellular calcium through phospholipase C, which subsequently potentiates BDNF release. 10 Therefore, both BDNF and DA have the potential to induce behavioral alterations observed in the central nervous system (CNS); however, the interaction between these two molecules in the cerebral cortex during nicotine exposure through CS is yet to be understood.
Amount and rate of absorption of nicotine into systemic circulation are crucial in determining the reinforcing effects of tobacco/nicotine in the smokers. The occupancy of nicotine at nicotinic acetylcholine receptors (nACh Rs) and its upregulation is induced by the chronic CS through nicotine’s interactions with the mesocorticolimbic DA system. 11 This may mediates nicotine’s positive reinforcing effects as well as cognition involving large-scale brain networks during task performance and at rest. 11 CS, like nicotine, indicates reinforcing effects for drug abuse, dependence, reward, and cognition during acute and chronic exposure. 12 Studies with the animal models and the human smokers on DA signaling in tobacco dependence further support the hypothesis that both the smoking behavior and nicotine administration result in the activation of the ventral striatum (VST) reward pathway via higher DA release and metabolism, and thus provide a common link of tobacco dependence with other behavioral addiction in human toward substance and nonsubstance agents. 13,14 DA transporter blockade in addiction model by psychostimulant also causes elevation of extracellular DA and downregulation of DA R, resulting in addictive behavior. 15
Moreover, various drugs and psychostimulants augment oxidative stress by producing reactive oxygen species (ROS) and reactive nitrogen species (RNS), depletion of cellular antioxidant defense, and subsequent cell death through either apoptosis or necrosis in diseases like, Parkinson’s and Alzheimer’s where DAergic system is involved. 16,17 BDNF level also decreases after exposure to oxidative stress in individuals with bipolar disorder, and modulates neuronal and behavioral plasticity, cognitive function etc. 18 Alterations in BDNF level are found to be associated with impairment of learning and memory in the rodents 19 , and in the schizophrenic patients. 20 Oxidative stress (i.e., ROS and RNS) has been associated with a wide range of DNA damaging events including base modification, strand breaking, and post-translational modifications of key proteins associated with development, neurotransmitter (NT) signaling, maturation, and synaptogenesis. 16,17 Further, high ROS and RNS are regulated in the cells by upregulation of COX-2 and inducible nitric oxide synthase (iNOS), respectively, that ultimately increases the susceptibility of cells toward degeneration and death contributing several disorders. 16,17 Nuclear factor-erythroid 2-related factor 2 (Nrf2) is the master regulator of cellular oxidative defense mechanism, which binds with antioxidant response element (ARE) in the promoter of several cytoprotective genes, including antioxidant and detoxifying enzymes, repair mediators, transcription factors of mitochondrial biogenesis, cell differentiation, and survival. 16,21 Nrf2 is also crucial for injury-induced neurogenesis in the hippocampus, regulating proliferation and differentiation of neural stem/progenitor cells. Possibly, Nrf2 may protect from mitochondrial dysfunction, and prevent from increase ROS generation and death of neural cells. In addition, the control of neuroinflammation by Nrf2 and the inhibition of proinflammatory factors like, iNOS or interleukin (IL)-6 may contribute to this effect. 21 Although Nrf2 is likely to be critical in pathogenesis of the adults’ cerebrum induced by oxyradical overload, but the involvement of Nrf2 and underlying NT mechanisms in such pathogenesis linked to behavioral alterations are yet to be answered.
Therefore, the present study focused on the potential toxic consequences of short-term (4 weeks) and long-term (12 weeks) exposure to nicotine/CS on the cerebral cortex of adult rats with regard to the neurobehavioral alterations. Here, our findings signify that nicotine/CS can induce ROS and RNS (through iNOS) with concomitant lowering of antioxidant defense (ascorbate and Nrf2) in the adult rat cerebral cortex. These changes, in turn, lead to breaking of double-stranded DNA (dsDNA), neurodegeneration of the cortex, and upregulation of BDNF, DA, and DA marker, TH, which are further associated with anxiogenic/depressive behaviors and cognitive impairments. Therefore, collectively, our data raise a possibility of novel therapeutic intervention targeted at neuropsychological disorders including cognition and drug addiction caused by tobacco smoking/nicotine.
Materials and methods
Chemicals and reagents
Chemicals and reagents were procured from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. Antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). TEMED, SDS, and tris-HCl were brought from HiMedia (Mumbai, India).
Animal treatment
Albino Wistar rats (250–275 g BW) were procured from Zydus Research Centre, Ahmedabad, and housed under standard conditions (12-hr dark/light cycles, 23 ± 3°C, 55 ± 15% RH) with food and water ad libitum. After one week of acclimatization, the rats were divided into control (Crt: n = 20), CS (n = 10) and nicotine (n = 30) groups (supplemental Table 1) for 4 and 12 weeks as per our previous protocol. 4 In the CS group, two commercially available filtered cigarettes (ITC, Kolkata, India) were passively inhaled for 1 hr/day in a tightly sealed whole body inhalation chamber as per our previous protocol. 22 In the control group, half of the rats (n = 10) were considered as untreated control for comparison with the CS group, and the remaining half (n = 10) were given vehicle (saline) ip for comparing with the nicotine group where nicotine (Nic 3–9 mg/kg BW/day) was dissolved in saline and administered orally. The doses were closely mimicking the amount of nicotine intake as with the CS in human; 23 hence, the animal model used here may closely resemble the human smoking situation. Further, human population is also exposed to chewing tobacco form, where route of nicotine entry is oral; therefore, in the present study, we compared both oral and smoking form of nicotine’s effects on the adult rat cerebral cortex. After behavioral experiments, the rats were anesthetized with ketamine + xylazine ip injection as mentioned previously, 22 and the cerebral cortex was dissected out. All procedures were performed in accordance with the guidelines from animal care and use committee of National Institute of Occupational Health (IAEC). Carcass was disposed by authorized private biomedical waste disposal agency.
Behavioral study
To analyze the neurobehavioral disorders, if any, under different experimental conditions as mentioned-above, individual rat was undergone anxiety/depression-related behavioral tasks for 5-min session in the elevated plus maze (EPM; Columbus instruments, Columbus, OH, USA). The rats were carried into the test room in their home cages and were placed in the central square of the maze facing the open arm by holding the base of their tails, and several behavioral scores were recorded as per the standard protocol 24 (supplemental Table 2). After each session, the rats were removed from the maze and returned to their home cage. The maze was then cleaned with 70% ethanol and permitted to dry between tests. Further, to determine the impairment of cognitive behaviors that acquired over time such as, learning, attention and memory, if any, upon exposure, Morris water maze (MWM) was performed. The rats were placed individually in a large water tank (dia × height: 140 × 35 cm) maintained at 27 ± 1°C just opposite (N ↔ S direction) to a hidden platform (9 cm dia, fixed 2 cm below the water surface) around the perimeter of tank facing toward the wall of the pool, and maximum 120-sec was given to perform the several behavioral scores as per Morris 25 (supplemental Table 3).
Sample preparation
Cerebral cortex homogenates were prepared in chilled 5 mM Tris-KCl buffer containing 0.1% triton X-100 (pH 7.4) followed by ultracentrifugation (12,000 rpm, 10 min × 2) to get the supernatant for oxidative stress and comet assay. For Western blotting, pooled cortex samples were lysed in buffer containing 10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% (v/v) NP40, 1 mM DTT, and 1 mM PMSF (pH 7.6), and nuclei were pelleted by 1000 rpm for 10 min. Supernatant was then ultracentrifuged at 14,000 rpm for 15 min to get the cytosolic fraction. The nuclear extract was prepared in buffer containing 20 mM HEPES, 420 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF, 0.5 mM DTT, and 25% (v/v) glycerol (pH 8.0), and centrifuged again at 1000 rpm for 10 min. All the buffers were supplemented with protease and phosphatase inhibitor cocktails. Samples were cryo-preserved for further analysis, if required.
Oxidative stress analysis
Total ROS was measured to detect the oxidative stress–induced damage of the cerebral cortex, if any, upon exposure, and the results were validated as mentioned previously. 22 Non-enzymatic antioxidant ascorbate was also measured by Roe and Kuether method. 26 Nrf2 and iNOS protein expressions were detected in the pooled cortex samples by Western blot. 22
Western blotting
To find out the alteration of the target protein expressions upon exposure, BDNF, TH, and iNOS in the cytosolic fraction and Nrf2 in the nuclear fraction were assayed through Western blotting as mentioned previously. 22,27 For this purpose, particular fractions containing 30 µg protein were loaded in SDS-PAGE and immunoblotting was done using different antibodies including β-actin as lane control to confirm uniform loading (supplemental Table 4). The protein of interest was visualized by Amersham ECL detection system (GE HealthCare Biosciences, Pittsburg, PA, USA) and densitometry of the protein was done by GelQuant.Net software (Biochemlabsolution.com, UCSF, Oakland, CA, USA).
Immunohistochemistry
To find out the localization of DA, TH, BDNF and Nrf2 in the cerebral cortex parallel to Western blot, immunohistochemistry (IHC) was performed on the paraffinized section using IHC kit (Vector Laboratories, Burlingame, CA, USA) as mentioned previously 22,28 (supplemental Table 4). Slides were observed in the bright field, and images were captured by Zeiss AxioScope microscope (Carl Zeiss Microscopy, GmbH, Göttingen, Germany) having attached digital camera at different magnifications. 22
Comet assay
To find out the oxidative stress–induced dsDNA breaking, if any, after exposure, we followed the modified alkaline comet assay (pH 13.0) in the cerebral cortex homogenate and the results were validated as per the standard protocol. 22,30 A total of 100 cells were analyzed on each slide to determine tail length, tail intensity, and ‘Olive tail moment’ by QWin software (Leica Microsystems, Gmbh, Wetzlar, Germany) and CASP software (http://www.casp.of.pl) using Leica DMI microscope (Leica Microsystems, Gmbh, Wetzlar, Germany) having attached with a computer. 22
Histology
To find out the oxidative stress–induced changes in the cerebral cortex histology after exposure, paraffin-embedded sections were used for the eosin–hematoxylin staining and photographed accordingly by Zeiss AxioScope microscope (Carl Zeiss Microscopy, GmbH, Göttingen, Germany) as per the previous protocols. 22,29
Neurodegeneration
After deparaffinization, 29 cerebral cortex sections were stained with fluoro-Jade C dye as per the vendor’s protocol (Chemicon, Temecula, CA, USA), which specifically determines neural cell death by neurodegeneration, and immunofluorescence (IF) images were taken by Leica DMI microscope (Leica Microsystems, Gmbh, Wetzlar, Germany) attached with a computer.
Imaging
Images of the same magnifications were considered between different groups, and good reproducible photomicrographs demonstrating obvious details were selected for publication.
Statistical analysis
One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test was performed using VassarStats.Net software (http://vassarstats.net). The results were considered significant at p < 0.05 and presented as mean ± standard error of mean (SEM) of the three independent experiments.
Results
Impact of nicotine and CS on weight, food and water consumption of adult rats
No significant difference in average body weight gain was observed in either of the exposed groups in case of both 4-week and 12-week treatment protocols, which is in corroboration with our earlier study. 4,22 Individual organ weights, food intakes, and water consumption were also not varied among the different study groups (data not shown) as reported earlier. 4,22
Nicotine- and CS-induced anxiogenic/depressive behavior in adult rats
Anxiety, stress, or depressive behavior among the adult rats due to the exposure of nicotine/CS was investigated by the EPM test. Both nicotine and CS at 4 and 12 weeks of exposure showed a similar strong preference for closed (Figure 1(b) and (e)) as opposed to open arms (Figure 1(a) and (d)) in the EPM. As compared to the respective controls, nicotine and CS caused significant defecation as a sign of anxiety/stress/fear in the dose- and time-dependent manner, the extent of which was quantified by the numbers of boli produced in the EPM during 5-min behavioral session for each rat in the respective experimental groups (Figure 1(c) and (f)). Further, no remarkable changes on the other spontaneous activities during EPM as anxiety/fear index of the rodents like, head dipping, rearing, locomotion, grooming and urination were observed (data not shown). But overall, the exposed rats exhibited enhanced reactivity to the anxiogenic stimuli, i.e., nicotine/CS, and engaged more time in self-grooming behavior (data not shown), a well-known behavioral response to anxiety eliciting situations. 31
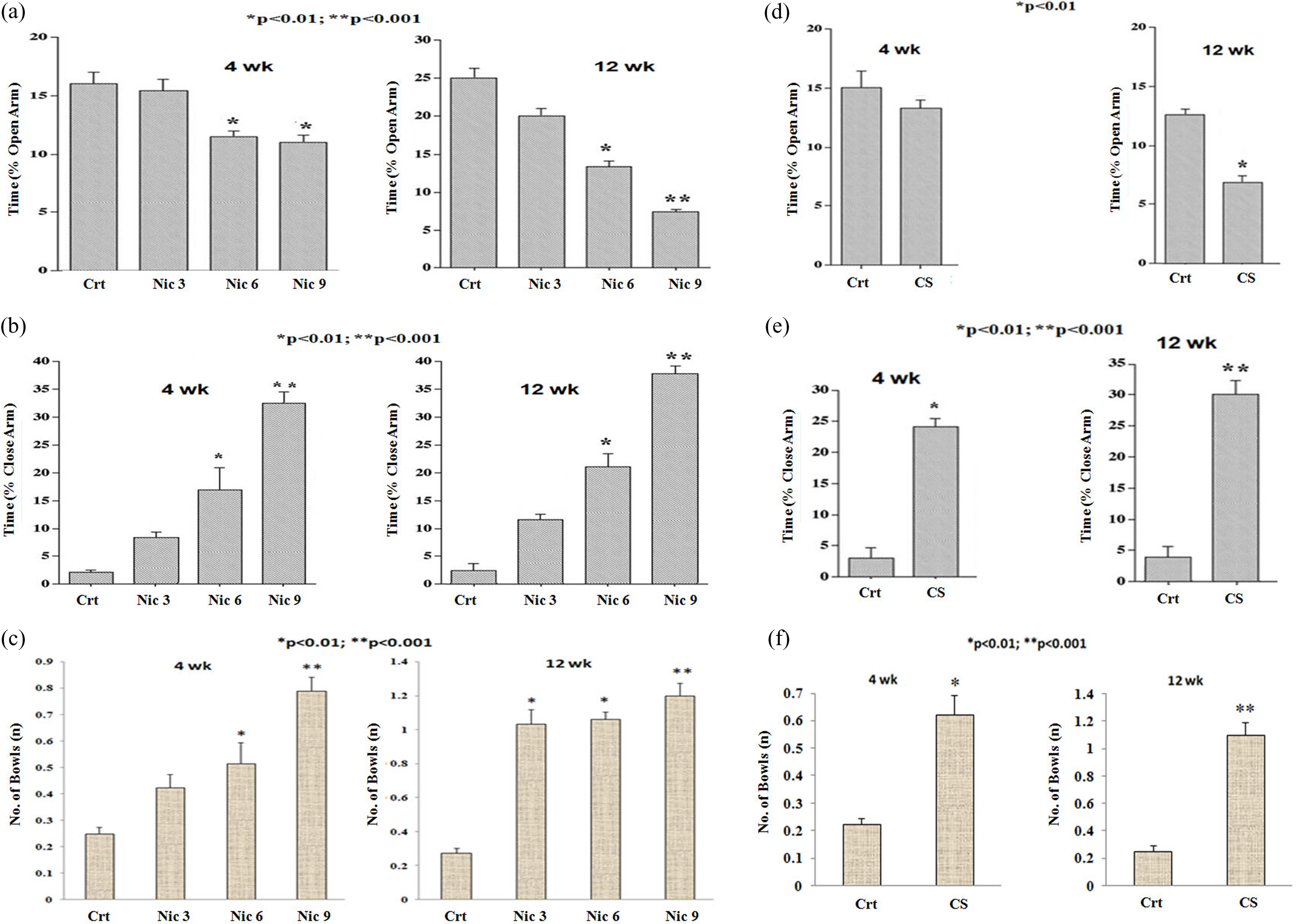
Effects of nicotine and CS on behavioral abnormality of the adult rats in EPM upon treatment with 3, 6, and 9 mg/kg BW/day nicotine (Nic, a-c) and 1 hr/day CS (d-f) for 4 and 12 weeks as compared to the respective controls (Crt). Detailed procedures are mentioned in Materials and Methods section. Results represent the mean ± SEM of the three independent experiments. *p < 0.01; **p < 0.001 (vs. Crt). CS: cigarette smoking; EPM: elevated plus maze.
Nicotine- and CS-inhibited cognitive and mood-associated behavior in adult rats
Cognitive behaviors include learning, memory and spatial working, and mood-associated behavior linked to the damage of the specific brain regions was assessed by the MWM tasks 25,32 following short- and long-term exposure of nicotine/CS. Compared to the respective controls, a significant increase in the escape latency time among the exposed groups (Figure 2(a) and (b)) was observed in the present study. Further, high exposed group spent more time at the maze periphery during swimming as escape behaviors after long-term exposure (12 weeks) of nicotine irrespective of the doses (Figure 2(a)) as well as CS (Figure 2(b)) than the respective controls. However, except escape latency, no other significant variation was observed for escape-like behavioral scores such as, climbing, diving, swimming pattern, swimming direction and path, limb movements, head angle and immobility of the rats while searching the escape route in the MWM (data not shown).

Effects of nicotine and CS on behavioral abnormality of the adult rats in MWM upon treatment with 3, 6, and 9 mg/kg BW/day nicotine (Nic, a) and 1 hr/day CS (b) for 4 and 12 weeks as compared to the respective controls (Crt). Detailed procedures are mentioned in the Materials and Methods section. Results represent the mean ± SEM of the three independent experiments. *p < 0.05 (vs. Crt). CS: cigarette smoking; MWM: Morris water maze.
Nicotine- and CS-induced neurodegenerative cell death and DNA damage in adult rats’ cerebral cortex
Cerebral cortex is linked with several behavioral outcomes and NT mechanism during drug/psychostimulant abuse and addiction. Therefore, to demonstrate whether the exposure to nicotine/CS may exert any negative impacts on the cerebral cortex correlates with the behavioral measures, we evaluated tissue histology, neurodegenerative cell death, and DNA comet. As shown in Figure 3(b) to (d), eosin–hematoxylin staining revealed dose-dependent structural changes such as, disintegration of the cells in the cerebral cortex region (from layer II onwards) of the adult rats upon short- and long-term exposure to nicotine/CS when compared to the respective controls (Figure 3(a)). Neurodegenerative cell death was remarkably noticed in the similar region of the cerebral cortex upon exposure (Figure 3(f) to (h)) by IF using a specific neurodegenerative fluorochrome and the CNS marker, Fluoro-Jade C. However, we could not find any change in the control cortex (Figure 3(e)). Further, to find out the DNA damaging potency of nicotine, comet assay was performed for determining the dsDNA breaking in the cerebral cortex (Figure 3(i) to (k)). The dose- and time-dependent increase in the percentage of comet tail DNA (i.e., tail intensity) along with the tail length was observed in the present study after nicotine/CS of 4 and 12 weeks as compared to the respective controls (Table 1). Further, higher DNA damage was noticed with high nicotine and CS for long-term exposure (12 weeks) than the low nicotine exposure for short term (4 weeks) as shown in Table 1. We also validated our data with the positive control (methyl methanesulfonate) and negative control (tris-buffer). As expected, methyl methanesulfonate induced DNA damage, whereas no DNA damage was observed after treatment with tris-buffer (Table 1).

Representative photomicrograph of the adult rats showing nicotine- and CS-induced cell disintegration neurodegeneration and DNA damage (DNA comet) in the cerebral cortex (↑) upon treatment with 6, and 9 mg/kg BW/day nicotine (Nic, c-d, g-h, k) and 1 hr/day CS (b, f, j) for 4 and 12 weeks as compared to the respective controls (Crt, a, e, i). Detailed procedures are mentioned in the Materials and Methods section. b, d, e-g: ×100; a, c, i-k: ×400; h: ×1000. CS: cigarette smoking.
Effect of nicotine and CS on DNA comet in the cerebral cortex of the adult rats upon treatment with 3, 6, and 9 mg/kg BW/day nicotine (Nic) and 1 h/day CS for 4 and 12 weeks as compared to the respective controls (Crt)a.
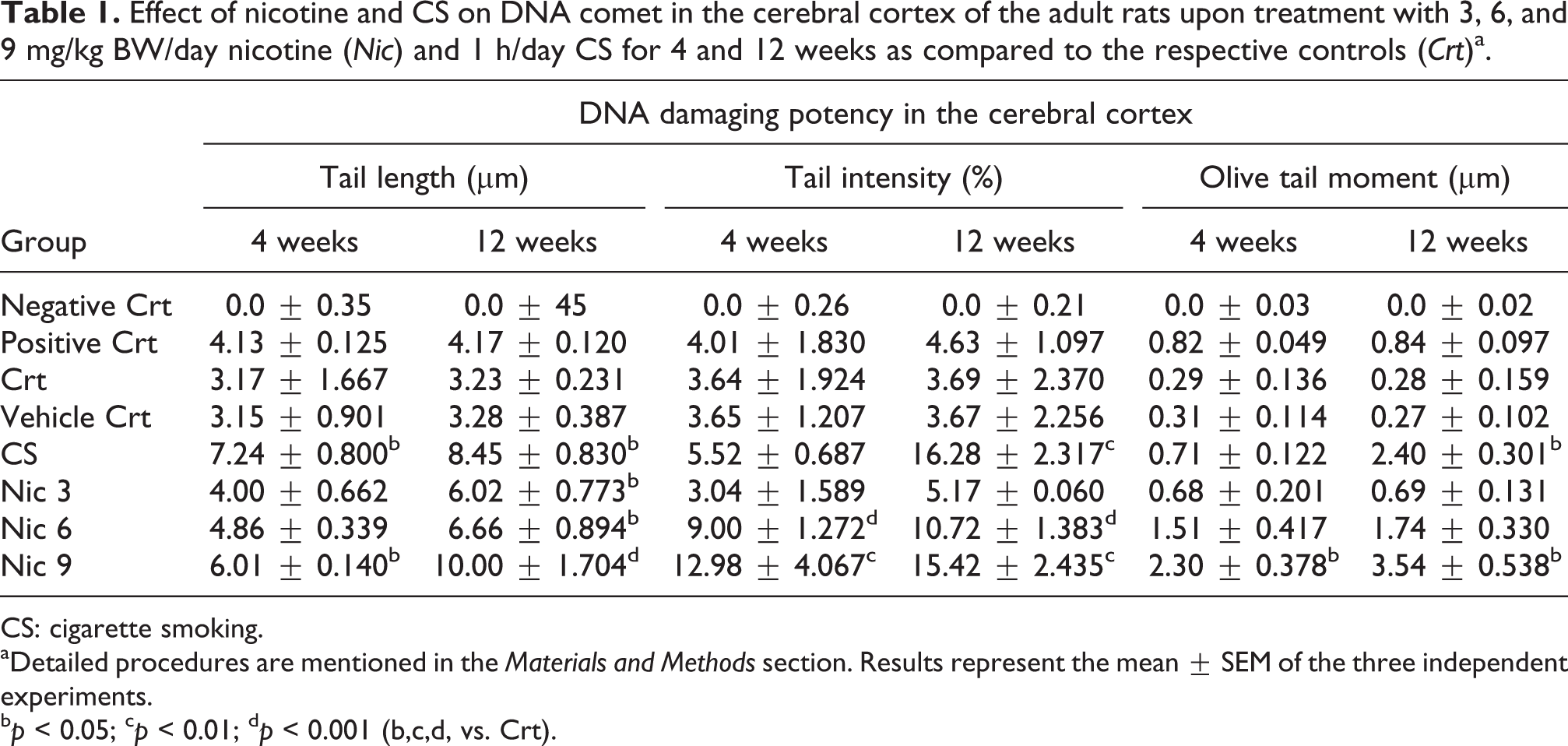
CS: cigarette smoking.
aDetailed procedures are mentioned in the Materials and Methods section. Results represent the mean ± SEM of the three independent experiments.
b p < 0.05; c p < 0.01; d p < 0.001 (b,c,d, vs. Crt).
Nicotine and CS inhibited antioxidant defense and induced oxidative stress in adult rats’ cerebral cortex
ROS and RNS have both beneficial and harmful roles, depending on their concentrations and duration of exposure. Therefore, to assess whether nicotine-/CS-induced cerebral cortical histopathology, DNA damage, and neurodegeneration are mediated through the alteration of cellular defense and oxidative mechanism, total ROS, iNOS, Nrf2, and ascorbate were measured under different experimental conditions as stated above. Compared with the respective controls, nicotine and CS dose-dependently showed significantly high level of ROS in the cerebral cortex upon 4 and 12 week treatment (Figure 4(a) to (b)). Figure 4(c) to (d) revealed significant reduction of ascorbate concentration in the cortex upon exposure to nicotine/CS for 4 and 12 weeks while comparing with the respective controls. Further, iNOS expression in the pooled cytosolic fraction of the cerebral cortex was upregulated remarkably with high concentration of nicotine/CS for 4 and 12 weeks of exposure in the adult Wistar rats (Figure 4(e) to (f)). The marker of cellular defense system, Nrf2, in the nuclear fraction of the same tissue showed significant dose- and time-dependent downregulation, whereas the controls revealed reciprocal relations in both the cases (Figure 4(e) and (g)); thereby establishing the dose- and time-dependent alterations of iNOS and Nrf2 protein expressions at the translational levels in the cerebral cortex induced by nicotine/CS. We also performed IHC in support of our Western blot data. Figure 4(i) to (k) indicated downregulation of Nrf2 in the same area of the cortex upon exposure to nicotine/CS as compared to the control (Figure 4(h)). Moreover, in the present study, upregulation of iNOS may be the causative factor of RNS production in the cerebral cortex after exposure.

Effects of nicotine and CS on oxidative stress and antioxidant defense of the adult rats upon treatment with 3, 6, and 9 mg/kg BW/day nicotine (Nic, a, c, e-g, i-j) and 1 hr/day CS (b, d, e-g, k) for 4 and 12 weeks as compared to the respective controls (Crt, a-h). Detailed procedures are mentioned in the Materials and Methods section. Results represent the mean ± SEM of the three independent experiments. The cortex homogenates are prepared for measurement of ROS by DCF-fluorescence (a-b) and ascorbate in mg/ml (c-d). $ p < 0.05; *p < 0.01; **p < 0.001 (vs. Crt). iNOS and Nrf2 proteins expression (e) in the pooled cortex homogenates are assessed by Western blot where β-actin is taken as loading control. Densitometric evaluations of iNOS (f) and Nrf2 (g) protein bands are done by GelQuant.Net software. $ p < 0.05; *p < 0.01 (vs. Crt). Representative photomicrographs of Nrf2 protein expression (brown) in the cerebral cortex region by IHC (h-k). k: ×100; h, j: ×200; i: ×400. CS: cigarette smoking; Nrf2: Nuclear factor-erythroid 2-related factor 2; iNOS: inducible nitric oxide synthase; ROS: reactive oxygen species.
We validated our data with the positive control (TPA), negative control (DMSO), and vitamin C–treated control. As expected, TPA significantly induced, whereas vitamin C alone significantly reduced the ROS concentration and iNOS expression in the control cortex, but the DMSO-treated sample remained unchanged (data not shown). Interestingly, downregulation of the TPA-mediated oxidative stress in the control cortex by vitamin C was also observed (data not shown). Taken together, our results indicated exposure to nicotine/CS caused inhibition of antioxidant defense by lowering of ascorbate and Nrf2 expression with concomitant induction of oxidative stress by increasing ROS and iNOS expression (therefore, RNS) in the adult rat cerebral cortex.
Nicotine and CS upregulated TH, DA and BDNF expressions in adult rats’ cerebral cortex
Finally, in order to determine the possible neurochemical targets of nicotine/CS-induced neurodegeneration, DNA damage, and oxyradical overload in the cerebral cortex of the adult rats, BDNF, DA and TH, the rate-limiting enzyme of DA biosynthesis, were evaluated by Western blot and IHC. As shown in Figure 5(a) to (c), TH and BDNF proteins were expressed significantly in the cytosol of the cerebral cortex upon exposure to nicotine/CS at 4 and 12 weeks as compared with the respective controls. Like Western blot, IHC also revealed intense co-localization of DA with BDNF and the DAergic marker, TH, in the same region of the cerebral cortex to CS/nicotine’s action (Figure 5(e) to (g)) while comparing with the control (Figure 5(d)). Overall, the dose- and time-dependent expression of BDNF, TH and DA proteins in the cerebral cortex suggested that nicotine/CS-induced upregulation of these neurochemicals at the translational levels occurs parallel with the behavioral alterations (measured by EPM and MWM), high oxidative stress (by ROS and iNOS), and low antioxidant defense system (ascorbate and Nrf2).

Effects of nicotine and CS on NT expression of the adult rats upon treatment with 3, 6, and 9 mg/kg BW/day nicotine (Nic, a-c, e) and 1 hr/day CS (a-c, f-g) for 4 and 12 weeks as compared to the respective controls (Crt, a-d). Detailed procedures are mentioned in the Materials and Methods section. Results represent the mean ± SEM of the three independent experiments. BDNF and TH (marker of DA) proteins expression (a) in the pooled cortex homogenates are assessed by Western blot where β-actin is taken as loading control. Densitometric evaluations of TH (c) and BDNF (b) protein bands are done by GelQuant.Net software. $ p < 0.05; *p < 0.01 (vs. Crt). Representative photomicrographs showed co-localization of BDNF (black), DA (brown) and TH (black) protein expression in the cerebral cortex region by IHC (d-g). d, f: ×100; e, g: ×400. CS: cigarette smoking: NT: neurotransmitter; TH: tyrosine hydroxylase; BDNF: brain-derived neurotrophic factor; iNOS: inducible nitric oxide synthase.
Discussion
In the present study, we, for the first time, provide evidences of significant cognitive and mood-associated behavioral alterations mediated by disruption of redox homeostasis, neurodegeneration, and irreversible oxidative modification of BDNF and DA expression in the cerebral cortex upon short-term (4 weeks) and long-term (12 weeks) exposure to psychostimulants nicotine and CS. Another issue relevant to this study is nicotine-, nornicotine- (produced from nicotine), and cotinine- (nicotine’s metabolic intermediate) induced release of DA through activation of nACh Rs. 33 CS and/or nicotine that generates during CS cause health hazards, exerting diverse physiological and biochemical effects. Ataxia, tremor, tachycardia, muscle fasciculation, and seizures were observed after administration of nicotine, in less than 5% rats twice or thrice out of the total 12-week treatment, which may be due to the initial phase of the nicotinic cholinergic stimulation as it is a parasympathomimetic alkaloid. 34 However, these signs and symptoms were disappeared within 2–3 min and no death was reported throughout the study period.
Previous studies found that explorative behavior in the rodents summarizes a broad spectrum of behavioral patterns such as, risk assessment behaviors, walking, rearing, climbing, sniffing and manipulating objects, which is gradually inhibited by anxiety; thereby representing an indirect measurement of anxiety. 35 Depression and anxiety spectrum disorders show considerable overlap in terms of diathesis and underlying neurobiology. 36 In conventional form of the test, anxiety is routinely assessed by the measurements of open arms avoidance. 37 We explored the performance of each rat in the anxiety-relevant paradigms through EPM based on the animal’s aversion in open arms, resulting in the avoidance of open arms (exploratory value) and confining movements to closed arms. Our data also revealed that long duration of nicotine/CS develops conflict between innate fear that the rodents have in open area vs. their desire to explore new environment. 38 As the natural tendency of the rodents is to prefer enclosed dark spaces as compared to open brightly light spaces; hence, by the degree to which the rodent avoids open arms of EPM in the present study, reflects a state of increased anxiety/fear/depression-related behaviors that corroborates previous observation. 37 Further, nicotine’s anxiolytic and anxiogenic effects depend on the age and sex of the animals. 38 Nevertheless, induction of anxiogenic behavior, depression, stress etc., was significantly higher with concomitant lowering of exploratory behavior upon high dose of nicotine/CS for longer duration of exposure in the present study, which indicates that the time spent is more important than the number of entries in such kind of the experimental set-up. Despite the lack of appropriate animal models to mirror the human mood condition, 39 our findings may have clinical implications as nicotine exerts negative effect, and the affected individuals may initiate smoking to regulate mood through a self-medication process, 40 whereas others would continue to smoke to manage mood dysregulation induced by their recreational exposure to nicotine itself. 41 Moreover, nicotine dependence is a complex phenomenon involving pharmacological and nonpharmacological factors mediated by the activation of the mesocorticolimbic DA system including VTA, NAcc, hippocampus, amygdale, and cerebral cortex. 11–12,42 Nicotine also increases glutamate release by agonistic actions at the excitatory presynaptic nACh Rs in these brain areas, which, in turn, modulates DA release in the projected terminal brain sites. 43
MWM study is indicative of learning and memory (spatial and working), and can also be used to assess the damage of the specific brain regions and cognitive behavior, if any, after pharmacological modification of drug abuse. 25,32 In MWM, the rodents locate a hidden platform using spatial cues surrounding the maze. 25 After carrying out the learning experiments in rats, some fail to perform water maze tasks and would simply float passively or remain immobile rather than swimming toward the escape platform, which is found to be reversed on the administration of antidepressants. 44 Studies also found that addicted tobacco users suffer from nicotine-induced cognitive and working memory impairments, mood disorders such as, stress, anxiety- and depression-like symptoms involving specific brain regions, especially the prefrontal cortex. 45 Simultaneously, nicotine dependence is also associated with the mood liability and anxiety, leading to increased feelings of stress in many regular smokers comparable to the nonsmokers, suggesting that acute nicotine withdrawal increases stress and nicotine reinstatement relieves the feelings of stress 46 through activation of the hypothalamus, cortex, amygdale and hippocampus. 47 Deficits in learning, memory and problem-solving skills are common in the children exposed to nicotine indicating that cognitive deficits link to the prenatal CS exposure. 41 Further, nicotine expression causes modifications in working memory; both ameliorations and impairments can occur due to even slight changes in the prefrontal DA levels in the human 48 and in the adult rodent offspring. 49 In the context of stress-induced behavioral changes and the influence of NTs, we demonstrated that impairment of cognitive memory (escape latency in MWM) and depression/anxiety-like behaviors (EPM tests) are induced by CS/nicotine in the adult rats. Also spending more time at the MWM periphery during swimming as escape behaviors of the adult rats upon long-term exposure in the present study is an indication of increased stress. It has already been reported that anxiety may increase thigmotaxis among the rodents, leading to impairments in searching the platform location in MWM possibly due to activity of the VST DA signaling. 50 Nicotine at low dose significantly improves consolidation and long-term memory, but has no effect on short-term memory following acute, subchronic and chronic treatments, suggesting that low dose nicotine generally improves memory, whereas no such effects are observed with high doses of nicotine. 51 In contrast, high dose of nicotine for 2-week treatment significantly increases escape latencies in MWM tests, but no such increase occurs with low dose as compared to the controls. Therefore, it has been indicated that high dose nicotine could impair learning and memory in the rats. 52 However, the present study revealed low, medium, and high doses of nicotine/CS as well as the duration of exposure are effective in impairing the MWM task (escape latency); hence, it can be suggested that both dose and time of exposure are equally important in such kind of study. Therefore, altogether, cognition may be limited to those cognitive processes that are acquired over time such as, learning, attention and memory, but not to the short-term memory influenced by nicotine.
Further, exposure to CS and nicotine causes structural changes in different brain regions like, apoptotic bodies in the hippocampus, 53 reduction in total cell number and DNA with hypertrophy of the remaining cells (therefore, increase total protein/DNA ratio) in the temporal cortex, 54 and alteration of DAergic system in the degenerative mid brain cPU. 4 Behavioral disorders may also be associated with such degenerative brain damage. 55 CS is an important environmental aging accelerator 56 partly because it induces oxidative stress and may contribute to many diseases including cognition- or neurodegeneration-related pathological changes in the brain. 57 Studies with animal models also reveal stress-related changes in neurogenesis and/or neurodegeneration of the several brain areas associated with mood, anxiety and depression. 58 DA, BDNF and oxidative burden prove to be the most stringent criteria for cellular integrity, structure–function impairment, total antioxidant depletion and NT function in the cerebral cortex. 16,18–19,59 Further, high oxidative stress induces alterations in antioxidant barrier within the CNS that could correlate with impairments in different brain structures, responsible for learning and memory processes. Nicotine activates hypothalamo–pituitary–adrenal axis via adrenocorticosteroid secretion; thereby increases blood cortisol level, responsible for the observed effects. 60 Overall, as compared to the controls, the dose- and time-dependent negative modulation of the exposed cerebral cortical histology and neurodegenerative cell death in the present study are found to be parallel with the EPM and MWM data.
Moreover, nicotine-induced DNA strand breakage is pH dependent and oxidative stress may be involved in this kind of DNA damage. 61 Thus, alkaline saliva generated by the chewing betel quid causes DNA damage, whereas high ROS contributes to oxidative stress–associated pathogenesis by disruption of the antioxidant balance, as vitamin C and E supplementations exert beneficial effects. 61 The enzymatic and nonenzymatic antioxidants, and the nutritional element of cellular system confer protection against oxyradical overload-induced tissue damage through scavenging action; hence, reduce stress burden and consequently improve anxiogenic, depressive, and other behavioral disorders. 16,21 Consistent with the previous observations, the dose- and time-dependent DNA damage in the cerebral cortex was noticed among the present exposed groups, which revealed high oxidative stress (ROS, iNOS) and antioxidant depletion (ascorbate, Nrf2) as compared to the controls.
Like ROS, RNS concentration also plays dual role in the various cellular processes including neurotoxicity, neurodegeneration, ischemia, tumor etc., depending on the activity and localization of the NOS isoforms in cells. 62 The enhanced expression of iNOS in the tobacco habituers compared to the nonhabituers indicates the possible influence of tobacco on iNOS expression and NO production. Ascorbate also acts as a neuronal modulator involving DAergic system linked to learning and memory processes. 63 Moreover, Nrf2 signaling pathway plays a central role in the regulation of cellular antioxidant mechanism on exposure to several environmental/oxidative stress, CS, nicotine, infection, or inflammation. 64–65 Nrf2, after exposure to oxidants, activates transcription of its target genes via ARE by disrupting the Nrf2-Keap1 interaction. 66 Studies also suggest that drugs that can activate Nrf2 are protective; hence, help in overcoming the negative impacts of coffee, tobacco and other psychostimulants. 67 The upregulation of a subset of putative Nrf2 target genes that regulate glutathione metabolism can also protect DA neurons. 68 Ramsey et al proved that oxidative stress could be an initiator or mediator of motor neuron death in diseases where nuclear translocation of Nrf2 is impaired, leading to dysfunction of the Nrf2/ARE pathway, resulting in decrease cellular defense system against oxyradical overload. 69 Therefore, combining data from our laboratory and elsewhere as mentioned-above, suggest that in the present study, (1) RNS production may be stimulated by the upregulation of iNOS expression in response to nicotine/CS, and (2) disbalance the redox homeostasis in the cerebral cortex microenvironment due to high RNS and ROS concentrations along with the downregulation of Nrf2. Moreover, consistent with its potent antioxidant activity, vitamin C alone antagonizes high ROS and RNS concentrations in the cerebral cortex, revealing vitamin C-induced blocking of the oxidative stress during our data validation. Finally, the observed histopathological and behavioral deficit of the cerebral cortex in the present study, mediated through the upregulation of ROS and RNS (by iNOS) with concomitant depletion of the antioxidant defense (i.e., ascorbate, Nrf2), is triggered by the dose- and time-dependent exposure of oral nicotine and passive CS to the adult Wistar rats.
In the present study, we further explore the underlying neurochemical mechanism in oxidative stress-induced degenerative changes of the cerebral cortex during nicotine/CS-induced cognitive and mood-associated behavioral disorders. Increasing evidence supports a role of NTs and neurogenesis in development of mood swifts although certain mediators may exert varying effects on different anxiety, depression and cognitive symptoms. Peri-adolescent stress produces several behavioral and neuronal abnormalities in the adult rats including increases BDNF and reduces DA D2R in the prefrontal cortex, and DA turnover in the NAcc, resulting in spatial learning deficit and latent inhibition. 70 Acute administration of nicotine increases firing rate of DAergic neurons in the VTA and elevates transmission of DA in the NAcc shell region through excitatory actions at nACh Rs on the same site and on the local neurocircuitary in different brain regions. 71 Thus, reinforcing effect of nicotine in the NAcc and striatum is characterized by increasing DA transmission that binds to DA Rs, which can further induce DA R downregulation. 5,15,33,71 Regulation of DAergic development is also mediated by other neurotrophic factors including BDNF, 9 which promotes sprouting of DAergic axons and regulates neuronal plasticity of DAergic neurons. 72 Nicotine also causes high BDNF levels in the hippocampus and cerebral cortex of the adult rats in either sex. 73 Previous studies also reveal that psychostimulant-induced enhancement of DA transmission 74 and TH at DAergic terminals mediated through NOS gene. 75 Moreover, NT-induced cell degeneration is thought to be involved in NT autoxidation and disruption of cellular redox system. 74,76 In mice, astrocyte-specific overexpression of Nrf2 protects DA neurons from toxins such as, 6-hydroxyDA that can elicit Parkinson’s-like neurodegenerative symptoms. 77 The smokers with mental disorders have significantly higher serum BDNF than the nonsmokers 20 linked with aging or weight gain, where peripheral BDNF levels may comprise of BDNF that originates from the CNS. 78 These observations suggest that nicotine-induced high level of peripheral BDNF may contribute to synaptic rearrangements involving development and maintenance of smoking habit as well as local activity in the CNS. Several studies further indicate nicotine-induced upregulation of BDNF and BDNF Rs in the midbrain DAergic system, 7 but downregulation of the hippocampal 5-hydroxytryptamine (5-HT) overflow. 79 It is possible, therefore, that a decrease in 5-HT R activation may be one mechanism by which long-term nicotine treatment increases BDNF expression in the brain. 80 Carlino et al reveal a positive correlation with truncated-BDNF in the schizophrenic individuals and worse performance in cognitive assays. 81 In consistent with the previous observations as discussed above, our data exhibited colocalization of BDNF, DA and DAergic component, TH proteins, in the cerebral cortical region (layer II onwards) upon exposure to nicotine/CS. Present findings also support that BDNF and NT, DA interventions as a possible mechanism of pro-oxidative stress (ROS, RNS, iNOS) in the CNS disorders during induction of psychostimulants, nicotine and CS.
Therefore, based on the results obtained so far, we may conclude that oral nicotine and passive CS exert irreversible secondary effect on BDNF-DA-Nrf2 signaling due to disbalance of redox homeostasis, which, in turn, degenerates cells in the cortex leading to poor cortical response, manifested by neurobehavioral abnormalities like, anxiogenic/depressive behavior and cognitive (learning/memory) alteration. It seems that nicotine and CS like other drugs and psychostimulants may suppress antiinflammatory factor, Nrf2, and upregulate proinflammatory factors, iNOS, COX-2, and so on 21 , which finally augment free radical- (ROS, RNS) induced cell death by neurodegeneration in the adult rat cerebral cortex. Because of Nrf2 downregulation, Nrf2-ARE mediated defense mechanism such as, activation of antioxidant enzymes, repair mediators, cell survival, behavioral gene etc., as well as neuroprotective activity of Nrf2 may no longer remain active. However, further mechanism-based studies on the activity of the R blockers in the animal models are warranted in order to elucidate the minute details of BDNF-DAergic-CAergic system and Nrf2 interaction with one another during development of neurobehavioral disorders, poor cortical response, and nicotine-dependence in tobacco abuse/withdrawal.
Supplemental material
Supplementary Material, Naha_Revised_Suppl_Cortex_final - Nicotine and cigarette smoke modulate Nrf2-BDNF-dopaminergic signal and neurobehavioral disorders in adult rat cerebral cortex#
Supplementary Material, Naha_Revised_Suppl_Cortex_final for Nicotine and cigarette smoke modulate Nrf2-BDNF-dopaminergic signal and neurobehavioral disorders in adult rat cerebral cortex# by Nibedita Naha, DN Gandhi, AK Gautam and J Ravi Prakash in Human & Experimental Toxicology
Footnotes
#
Acknowledgement
This work was approved by Institute Animal Ethical Committee (IAEC/NIOH/2013-2014/01) and supported by the Intramural Research Program of ICMR-NIOH, Govt. of India. The authors thank Director-in-Charge, ICMR-NIOH for providing infrastructural facility including animal house (Dr. R Palkhade). Thanks are also given to Prof. PP Nayak, NRI Medical College & General Hospital, Guntur, and Dr. S Mondal, ICAR-National Institute of Animal Nutrition & Physiology, Bangalore, for manuscript editing.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research of this article: This research was supported by the Intramural Research Program of ICMR-NIOH, Govt. of India.
Supplemental material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.