
Abstract
Cigarette smoke (CS) is a major risk factor for emphysema, which causes cell death in structural cells of the lung by mechanisms that are still not completely understood. We demonstrated previously that CS extract (CSE) induces caspase activation in MRC-5 human lung fibroblasts, activated protein kinase C-η (PKC-η), and translocated PKC-η from the cytosol to the membrane. The objective of this study was to investigate the involvement of PKC-η activation in a CSE-induced extrinsic apoptotic pathway. We determined that CSE increases expression of caspase 3 and 8 cleavage in MRC-5 cells and overexpression of PKC-η significantly increased expression of caspase 3 and 8 cleavage compared with control LacZ-infected cells. In contrast, dominant negative (dn) PKC-η inhibited apoptosis in MRC-5 cells exposed to CSE and decreased expression of caspase 3 and 8 compared with control cells. Exposure to 10% CSE for >8 h significantly increased lactate dehydrogenase release in PKC-η-infected cells compared with LacZ-infected cells. Additionally, PKC-η-infected cells had an increased number of Hoechst 33342 stained nuclei compared with LacZ-infected cells, while dn PKC-η-infected cells exhibited fewer morphological changes than LacZ-infected cells under phase-contrast microscopy. In conclusion, PKC-η activation plays a pro-apoptotic role in CSE-induced extrinsic apoptotic pathway in MRC-5 cells. These results suggest that modulation of PKC-η may be a useful tool for regulating the extrinsic apoptosis of MRC-5 cells by CSE and may have therapeutic potential in the treatment of CS-induced lung injury.
Introduction
Chronic obstructive pulmonary disease (COPD) is associated with chronic inflammation of the peripheral airways and progressive destruction of the lungs, which leads to gradual obstruction of the airways. 1,2 The pathogenesis of COPD remains poorly understood, although it may involve lung inflammation caused primarily by cigarette smoke (CS) associated with protease/anti-protease imbalance and oxidative stress. 1,3 Recent data suggest that programmed cell death is important to the pathogenesis of COPD, interacting with inflammation and protease/anti-protease imbalance, and contributing to the destruction of lung parenchymal tissue in response to CS. 4,5
Several studies have reported apoptosis and apoptogenic factors induced by CS in vivo, as well as in a variety of cultured human cells, including the monocytic cell line U-937, human lung fibroblast, and umbilical vein endothelial cells. 6–9 The precise molecular mechanisms underlying the apoptotic pathways triggered and regulated by CS are still not completely understood in terms of COPD pathogenesis. CS extract (CSE) induces intrinsic apoptosis in alveolar macrophages by increasing reactive oxygen species generation, mitochondrial dysfunction, Bax accumulation, and cytochrome c release, independent of p53, Fas, or caspase activation. 8 Recently, several studies have reported that the level of the pro-apoptotic proteins caspase 3, caspase 8, Bax, truncated Bid, and cytochrome c increased in lung specimens of CS-exposed rats, 10,11 indicating that CS sensitized lung cells toward apoptosis involving elements of both extrinsic and intrinsic pathways.
Protein kinase C (PKC) family consists of 12 related serine/threonine protein kinases, some of which are known as critical regulators of cell proliferation, cell survival, and cell death. PKCs have been divided into three subgroups based on their regulation, namely conventional PKCs (α, ßI, ßII, and γ), novel PKCs (δ, ε, θ, μ, and η), and atypical PKCs (ζ, λ, and τ). Each PKC isoform is expressed by an individual gene, except for βI and βII, in a tissue-specific manner and responds to activation by distinct stimuli. Differential roles of PKC have been reported depending on specific isoforms, cell type, and/or different apoptotic stimuli. Currently, the role(s) played by PKC in CSE-induced apoptosis is unclear. In Fas receptor-mediated apoptosis, Fas interaction with Fas-associated death domain protein and caspase 8 are involved in formation of the death-inducing signaling complex (DISC). 12 A previous study reported that CSE induces Fas receptor-mediated DISC formation involving extrinsic apoptosis, 9 which is differentially regulated by PKCα and PKCζ via the phosphatidyl nositol 3-kinase (PI3K)/Akt pathway.
In this study, we used MRC-5 human lung fibroblasts to study the involvement of PKC-η, a novel PKC isoform, in extrinsic apoptosis induced by CSE.
Materials and methods
Adenovirus-mediated gene expression
Replication-deficient Ad5-type adenovirus vectors containing the complementary DNA of both wild-type (wt) and kinase-negative mutants of rabbit PKC-η were constructed as described previously. 13
β-Galactosidase assay
β-Galactosidase activity was measured using a mammalian β-galactosidase assay kit (Thermo-Pierce, Rockford, Illinois, USA) according to the manufacturer’s protocol. In brief, cells were washed once with phosphate-buffered saline (PBS) buffer, and a sufficient volume of mammalian protein extraction reagent was added to each well. After incubation for 10 min at room temperature, cells were scraped from the dish and centrifuged at top speed for 15 min at 4°C. An amount of β-galactosidase assay reagent equal to the volume of lysate remaining was then added to each well. After incubation at 37°C for 30 min, the reaction was stopped by adding 150 µl stop solution. The absorbance of each well was measured at 405 nm using a microplate reader (Molecular Devices Emax, Sunnyvale, California, USA).
Preparation of CSE
Kentucky 3R4F research reference-filtered cigarettes (The Tobacco Research Institute, University of Kentucky, Lexington, Kentucky, USA) were consumed using a peristaltic pump (VWR International, Radnor, Pennsylvania, USA). Before the experiments, the filters were cut from the cigarettes. Each cigarette was consumed in 6 min with a 17-mm butt remaining. Eight cigarettes were bubbled through 40 ml of cell growth medium, and this solution, regarded as 100% strength CSE, was adjusted to a pH of 7.45 and used within 15 min after preparation.
Cell culture and treatments
The MRC-5 human lung fibroblast cell line was purchased from the American Type Tissue Collection (Manassas, Virginia, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Grand Island, New York, USA) supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% carbon dioxide. For adenoviral infections, cells were grown to 40% confluence and fresh growth medium containing 109 colony-forming units (CFU)/ml of adenoviral vector expression PKC-η, dominant negative (dn) PKC-η, or LacZ. Infected cells were incubated for 18 h and then washed in serum-free medium. For CSE treatment, the cells were exposed to 10% CSE at indicated time intervals.
Cytotoxicity and viability assays
Lactate dehydrogenase (LDH) release was measured using a cytotoxicity detection kit (Roche, Indianapolis, Indiana, USA) according to the manufacturer’s protocol. After gentle agitation, 100 μl of culture medium was collected at various times during the assay. Cell viability was measured with blue formazan, metabolized from 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) by mitochondrial dehydrogenases that are active only in live cells. MRC-5 cells were seeded in 48 well plates (5 × 103 cells/well) and incubated in DMEM for 24 h. Cells were exposed to 10% CSE for the indicated time intervals, MTT reagent (5 mg/ml) was added to each well, and then the plate was incubated for an additional 2 h at 37°C. The medium was then removed, and the intracellular formazan product was dissolved in 300 μl dimethylsulfoxide. The absorbance of each well was measured at 540 nm with correction at 650 nm using a microplate reader (Molecular Devices Emax). Optical density values from untreated control cells were designated 100% as a standard.
Nuclear staining with Hoechst 33342
Apoptosis was examined by staining the cells with Hoechst 33342 (Sigma, St Louis, Missouri, USA). MRC-5 cells were washed twice with PBS and then fixed in PBS-containing 4% formaldehyde for 10 min at room temperature. Fixed cells were washed with PBS and stained with Hoechst 33342 for 5 min at room temperature. Cells were evaluated under a fluorescence microscope (OlympusIX70; Olympus Corp., Tokyo, Japan) for nuclei exhibiting typical apoptotic features, such as chromatin condensation and fragmentation. Photographs were taken at a magnification of 200×.
Caspase activity assays
The activities of caspases 3 and 8 were determined by a colorimetric assay using kits from R&D System (Wiesbaden-Nordenstadt, Germany), according to the manufacturer’s protocol. Briefly, cells were lysed in the supplied lysis buffer and were incubated on ice for 10 min. At the end of incubation, cell lysates were centrifuged at 10,000g for 10 min at 4°C to precipitate cellular debris. The supernatants were collected and incubated with the supplied reaction buffer containing dithiothreitol and Asp-Glu-Val-Asp-p-nitroaniline (DEVD-pNA) (specific for caspase 3) or Ile-Glu-Thr-Asp-p-nitroaniline (IETD-pNA) (specific for caspase 8) as substrates at 37°C. The reaction was measured by absorbance at 405 nm using a microplate reader (Thermo Scientific, Waltham, Massachusetts, USA). Enzyme activity was expressed as the nth-fold increase in the proportion of apoptotic cells over that of nontreated control cells.
Subcellular fractionation and Western blotting
Cells were lysed in hypotonic buffer (20 mM; 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.4), 1.5 mM magnesium chloride, 10 mM potassium chloride, and protein inhibitor cocktail). The lysates were passed through a 25-gauge needle 10 times using a 1-ml syringe and left on ice for 20 min. After incubation, lysates were centrifuged 100,000g for 1 h at 4°C, yielding the supernatant as a soluble (cytosolic) fraction. The pellet was resuspended in 1% Triton X-100-containing buffer and centrifuged 10,000g for 20 min at 4°C. The supernatant was recovered as a membrane fraction. Prior to polyacrylamide gel electrophoresis, lysates were boiled for 10 min, followed by centrifugation at 13,000 r/min for 5 min at 4°C. They were separated via 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene diflouride membrane (Millipore, Bedford, Massachusetts, USA). The membrane was blocked with 5% (w/v) nonfat milk powder in tris(hydroxymethyl)aminomethane (Tris)-buffered saline and Tween 20 mixture (TBST; 50 mM Tris-hydrochloric acid, 150 mM sodium chloride, 0.1% Tween-20) for 1 h at room temperature. After three washes in TBST, the membranes were incubated with primary antibodies (cleaved caspase 3 and cleaved caspase 8, Cell Signaling, Boston, Massachusetts, USA, and PKC-η, Santa Cruz Biotechnology, Dallas, Texas, USA) at a dilution of 1:500 in TBST-containing 5% nonfat milk overnight at 4°C. After three washes in TBST, the membranes were incubated for 1 h at room temperature with the horseradish peroxidase-conjugated secondary antibody at a 1:5000 dilution. Bound antibodies were detected using an enhanced chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK) following the manufacturer’s instructions. The loading amount in each well was normalized using an anti-actin antibody. Protein concentration was determined using the bicinchoninic acid assay (Intron, Seoul, Korea).
Statistical analysis
All values are expressed as means ± SE. Statistical significance was determined by Student’s t test, and a value of p < 0.05 was considered significant.
Results
Cell viability and cytotoxicity in MRC-5 cells after CSE exposure
We evaluated the effect of CSE on MRC-5 cell death by monitoring LDH release. MRC-5 cells were exposed to 0, 1, 5, and 10% (v/v) CSE at the indicated times. As shown in Figure 1(a), LDH release was significantly increased in a CSE dose-dependent manner relative to untreated cells. Cell viability was measured with the same treatments using the MTT assay. Cell viability after CSE exposure was decreased significantly in a time-dependent manner (Figure 1(b)). Therefore, we conducted a further investigation into the CSE concentration and kinetic combinations that preceded the appearance of significant LDH release or loss of viability.

CSE induces cell death in human lung fibroblasts. MRC-5 cells were exposed to 0, 1, 5, and 10% (v/v) CSE at the indicated times. Cell cytotoxicity was assessed using an (a) LDH assay and (b) MTT assay. Data from each time point were compared with untreated cells at the same time. The data represent means ± SD of three independent experiments. *p < 0.05; ***p < 0.001: compared with untreated cells. CSE: cigarette smoke extract; LDH: lactate dehydrogenase; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
CSE induces expression of caspase 3, caspase 8, and PKC-η in MRC-5 cells
We evaluated the activation of caspase 8 as a function of CSE exposure, because the proteolytic autoactivation of caspase 8 activates downstream caspase 1 and caspase 3. Caspase 3 is an executioner caspase, whose activation represents a distal event in the apoptosis signaling pathway. Activation of caspase 3 or caspase 8 was assessed by Western blot. Caspase 3 or caspase 8 is normally present in an inactive proenzyme form but can be activated by proteolytic processing of its inactive zymogen into its cleaved p17, p19 forms and p18, p26 forms, respectively. After exposing MRC-5 cells to 10% CSE at the indicated times, we observed increased expression of the cleaved caspase 3 subunit p19 and the cleaved caspase 8 subunit p26 after 18 h (Figure 2(a)). These results indicate that CSE exposure induces MRC-5 cell death through the apoptotic pathway. We also investigated whether CSE can alter the expression or activation of PKC isoforms in MRC-5 cells by Western blotting of cell lysates. Total expression of PKC-η was not altered in response to 10% CSE (Figure 2(b)), but expression levels increased in PKC-η in a time-dependent manner. To evaluate PKC activation, MRC-5 cells treated with 10% CSE for 0, 1, 3, and 6 h were fractionated to isolate membrane and cytosolic fractions, as described in the Materials and Methods section. Both fractions were prepared for Western blot analysis, and the results indicated that the membrane fraction of MRC-5 exhibited increased PKC-η after 1 h of CSE exposure (Figure 2(c)).
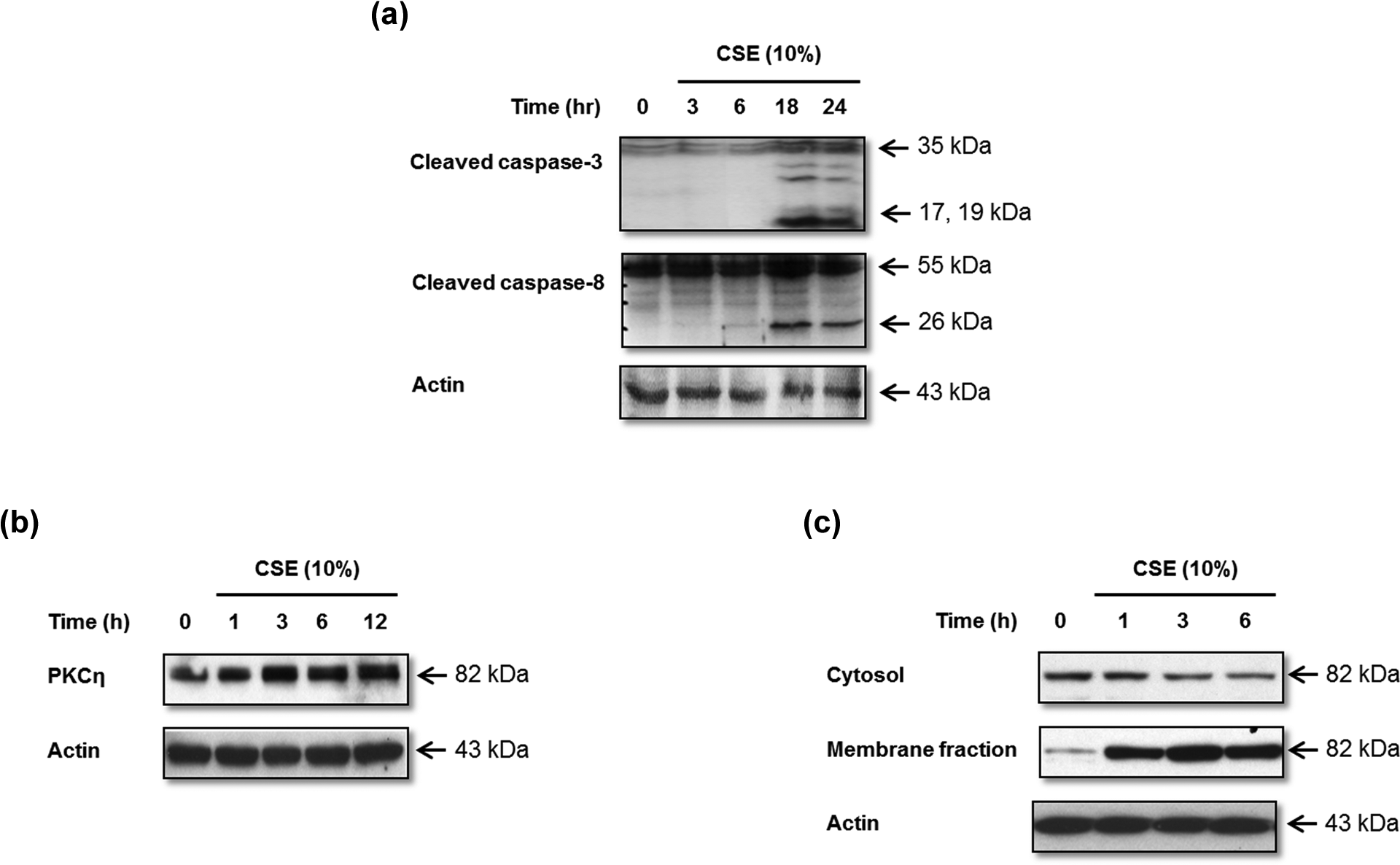
CSE induces caspase activation and PKC-η expression in human lung fibroblasts. MRC-5 cells were treated for the indicated times with 10% CSE and Western blots performed to assess (a) activation of caspases 3 and 8, (b) PKC-η in total cell lysates, and (c) translocation of PKC-η from the cytosol to the membrane fraction. Actin was used as a loading control. CSE: cigarette smoke extract; PKC: protein kinase C.
PKC-η regulates CSE-induced caspases 3 and 8 activity in human lung fibroblasts
A previous study demonstrated that CSE induces MRC-5 cell death through the extrinsic apoptotic pathway. 9 Hence, we further investigated the apoptotic pathway involved in CSE-induced PKC-η activation by focusing on the extrinsic pathway. Because activation of caspase-8 represents an initial event in extrinsic apoptotic signaling, we assessed caspases 3 and 8 activities using Western blot and a colorimetric assay. MRC-5 cells were infected with PKC-η, dn PKC-η, or LacZ and then exposed to 10% CSE for 18 h. The activation of caspases 3 and 8 following CSE exposure increased in PKC-η-infected cells, but decreased in dn PKC-η-infected cells, compared with LacZ-infected cells (Figure 3(a) to (d)). The efficiency of adenoviral infection was assessed in LacZ-infected cells by monitoring β-galactosidase activity (Figure 3(e)). These data indicate that PKC-η activates apoptosis via a caspase-dependent apoptotic cascade induced by CSE.
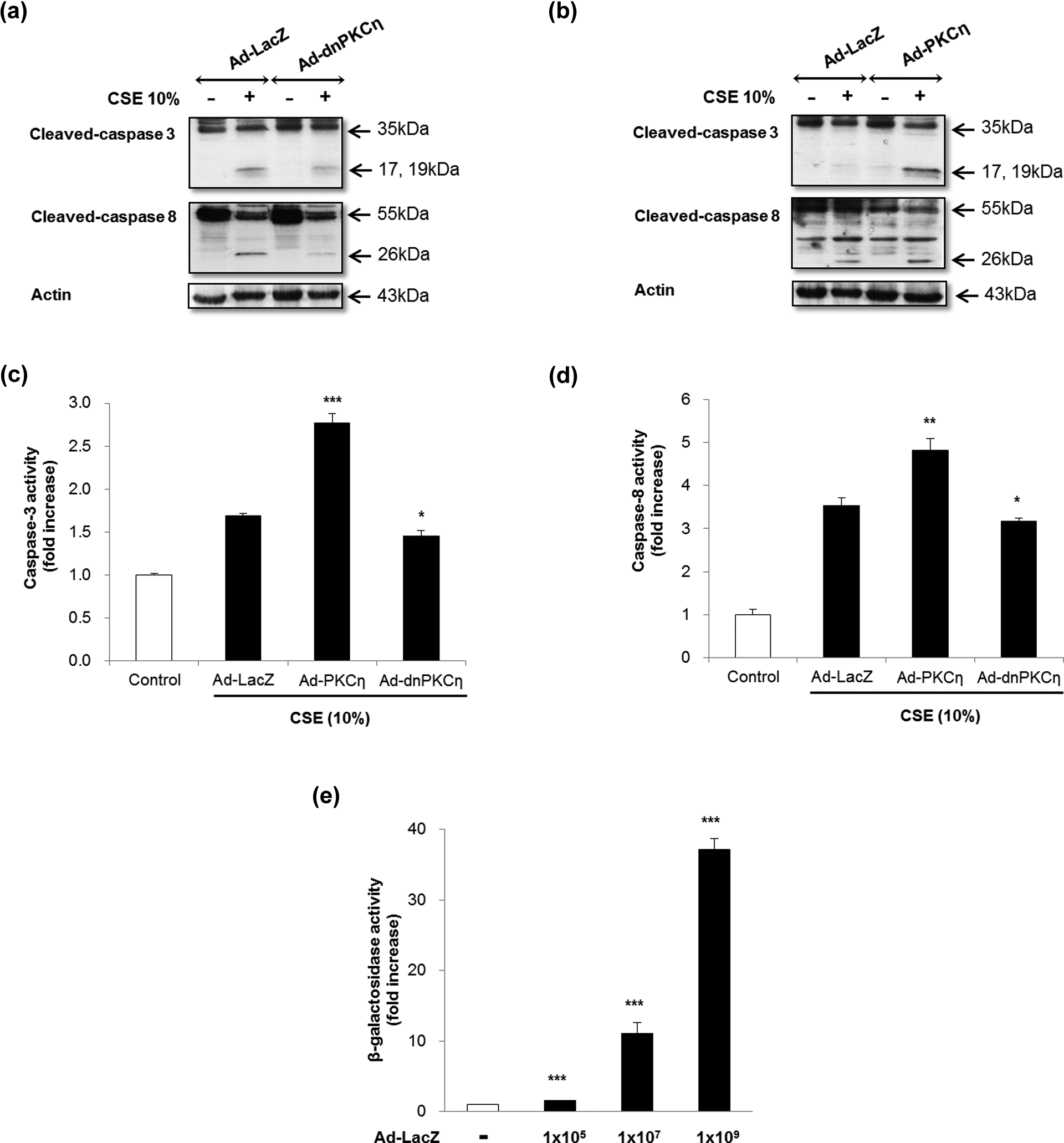
PKC-η increases caspases 3 and 8 activity in human lung fibroblasts. MRC-5 cells were infected with 109 CFU/ml of dn PKC-η or PKC-η or LacZ-expressing adenovirus in culture medium and incubated for 18 h prior to CSE exposure. Protein expression of cleaved caspases 3 and 8 was assessed by Western blotting in (a) LacZ and dn PKC-η-infected cells and (b) LacZ and PKC-η-infected cells. Actin was used as loading control. (b) Caspase 3 activity was assessed in PKC-η and dn PKC-η-infected cells and compared with LacZ. (d) Caspase 8 activity was assessed in PKC-η and dn PKC-η-infected cells and compared with LacZ. (e) The efficiency of adenoviral infection was determined in LacZ-infected cells by monitoring β-galactosidase activity. The data in (c) to (e) represent means ± SD of three independent experiments. *p: <0.05; **p: <0.01; ***p: <0.001; PKC-η: protein kinase C-η; dn PKC-η dominant negative protein kinase C-η; CSE: cigarette smoke extract.
PKC-η regulates CSE-induced cell death in MRC-5
We investigated whether the activation of PKC-η in CSE-induced cell death is an apoptosis-mediated effect using an LDH assay. MRC-5 cells were infected with PKC-η, dn PKC-η, or LacZ and then exposed to 10% CSE for 8 h. As shown in Figure 4(a), the rates of LDH release of PKC-η, dn PKC-η, and LacZ-infected cells were 2.32-, 1.85-, and 2.07-fold, respectively. Cell death was significantly increased in PKC-η-infected cells compared with LacZ-infected cells (p < 0.01). We also performed Hoechst 33342 staining to assess morphological changes. Chromosomal condensation and nuclear fragmentation were observed in many MRC-5 cells exposed to 10% CSE for 18 h (data not shown). PKC-η-infected cells increased the number of Hoechst 33342 stained nuclei compared with LacZ-infected cells (Figure 4(b), top). However, dn PKC-η-infected cells exhibited fewer morphological changes than LacZ-infected cells under phase-contrast microscopy (Figure 4(b), bottom). These results indicate that wt PKC-η activates CSE-induced apoptotic cell death.

PKC-η promotes CSE-induced cell death in human lung fibroblasts. MRC-5 cells were infected with 109 CFU/ml of dn PKC-η or PKC-η or LacZ-expressing adenovirus in culture medium and incubated for 18 h prior to CSE exposure. (a) An LDH assay was used to evaluate the cytotoxicity after 8 h incubation with 10% CSE. Data from PKC-η and dn PKC-η-infected cells were compared with LacZ. The data represent means ± SD of three independent experiments. (b) Nuclei of MRC-5 cells were stained with Hoechst 33342 to detect apoptosis morphologically. Images from phase-contrast microscopy are also displayed (×200). *p: <0.05; **p: <0.01; PKC-η: protein kinase C-η; CSE: cigarette smoke extract; dn PKC-η: dominant negative protein kinase C-η; LDH: lactate dehydrogenase.
Discussion
Although the role of apoptosis in CSE-induced cell death is still not completely understood, 14,15 we demonstrated that CSE exposure induced an extrinsic apoptotic pathway involving activation of caspases 3 and 8 in MRC-5 cells (Figure 2(a)). The relative role of individual PKC isoforms in Fas-mediated apoptotic pathway and caspase activation remains unclear. The expression or activation states of PKCs in response to CSE exposure and how they affect CSE-induced apoptosis also remain unclear. CSE induces preferential translocation of PKC-α and -ζ isoforms from the cytosol to the plasma membrane, which is necessary for activation. 9 In this study, we demonstrated that PKC-η is activated by CSE (Figure 2(b) and (c)). A previous study demonstrated that PKC-α and -ζ differentially regulates DISC formation and are involved in CSE-induced apoptosis. The mechanism(s) by which PKC-α and -ζ regulate DISC formation during CSE-induced apoptosis remain to be clarified; however, our findings that the introduction of dn PKC-α reduces Akt phosphorylation, whereas dn PKC-ζ increases Akt phosphorylation, suggesting a potential role for the pro-survival PI3K/Akt pathway. 9
The biological roles played by PKC-η in the regulation of human lung fibroblastic cell death induced by CSE have not been well characterized. Although some researchers have suggested that PKC-η plays a pro-apoptotic role in the regulation of cell division and cell death during early B-cell development, 16 PKC-η also reportedly blocks the apoptotic cascade via caspase 9 activation following ultraviolet (UV) and gamma irradiation-induced apoptosis in glioblastoma cells 17 and UV-induced activation of caspase 3 in normal human keratinocytes. 18 Downregulation of PKC-η rendered a Hodgkin’s lymphoma-derived cell line, more sensitive to doxorubicin and camptothecin. Furthermore, knockdown of PKC-η by small interfering RNA revealed increased poly adenosine diphosphate ribose polymerase (PARP)-1 cleavage, cytochrome c release, and caspase 7 activation. 19 These results differ from our present findings documenting the protective role of PKC-η inhibitor against CSE-induced MRC-5 cell death. In this study, LDH release was increased in PKC-η-infected cells, but decreased in dn PKC-η-infected cells (Figure 4(a)). We also demonstrated that the expression of caspases 3 and 8 cleavage was decreased in dn PKC-η-infected cells after CSE exposure (Figure 3(a)). The present results are sufficient to explain the pro-apoptotic role of PKC-η, because PKC-η increased the expression of caspases 3 and 8 (Figure 3(b)). In contrast, inhibition of PKC-η resulted in anti-apoptotic effects in CSE-exposed MRC-5 cells, measured by following caspase activity (Figure 3(c) and (d)). In other studies, overexpression of a constitutively active form of PKC-η played a role in stimulating c-Jun N-terminal kinase (JNK) activation in human embryonic kidney 293 cells. In contrast, inducible PKC-η expression in MCF-7 cells inhibited JNK activity and apoptosis in response to DNA damage. 20 The molecular mechanisms underlying the regulation of JNK/c-Jun signaling by PKC-η may be dependent on physiological conditions or be cell-type specific. Therefore, we suggest that the functional role of PKC-η is complicated and that this enzyme may exert either pro- or anti-apoptotic effects depending on the cell type and/or type of injury. Different results between cells and insults may, in part, help explain this discrepancy in apoptosis and differentiation; therefore, further study is required to delineate the precise role of PKC-η using various PKC-η activators and inhibitors in a CSE-induced cell death model. In this study, we selected fibroblasts as one cell type to perform a detailed mechanistic study of apoptosis by CSE. The lung is structured as a heterogeneous organ consisting of multiple cell types including fibroblasts of the lung parenchyma, as well as bronchial, airway, and alveolar epithelial cells, endothelial and smooth muscle cells of the pulmonary vasculature, smooth muscles of the airway, alveolar macrophages, and other specialized cells such as mast cells. We cannot exclude the possibility that apoptotic responses in other lung cell types may have an equal or perhaps greater contribution to the pathogenesis of pulmonary diseases triggered by CS. A previous study demonstrated that CS induced DISC formation and caspase 8 activation in human bronchial epithelial cells (Beas-2B), which is consistent with the fibroblast results. 21
This study is the first to demonstrate that overexpression of PKC-η increases CSE-induced caspases 3 and 8 activities, and subsequently cell death, while dn PKC-η attenuates CSE-induced apoptosis. Taken together with previous PKC-α and -ζ data, our present results suggest that PKC-η functions as a pro-apoptotic protein in MRC-5 cell lines, and modulating compounds may be useful tools for regulating apoptosis and for clarifying PKC-η activation in cell death pathways induced by CSE.
Footnotes
Funding
This work was supported by Research Grants funded by Gachon University Gil Medical Center in 2011 to JW Park.