
Abstract
(R)-Goniothalamin (R-GNT) is a secondary metabolite isolated from the plants of the genus Goniothalamus. This molecule has attracted the attention of researchers because of its selective cytotoxicity against tumor cells and its ability to induce apoptosis. (S)-Goniothalamin (S-GNT) is a synthetic enantiomer of R-GNT, and its mechanism of action is largely unknown. In this study, we investigated the activity of S-GNT in a human non-small cell lung cancer NCI-H460 cells. We observed that the cells exposed to this compound exhibited cytotoxicity in a concentration-dependent manner. Based on the data obtained through the assessment of apoptosis induction in situ and the comet assay, we suggest that this cytotoxicity occurs due to the potential ability of this molecule to induce DNA damage with the consequent induction of cell death via apoptosis. A significant reduction in the messenger RNA levels of baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5) gene that encodes the survivin protein was found. This novel finding may explain the inhibition of cell proliferation and induction of apoptosis in tumor cells caused by this compound.
Introduction
Since 1980s, lung cancer has been the most common type of cancer in the world. Currently, this disease has some of the highest reported incidence and mortality rates among cancers. In the last world estimate, approximately 1.61 million cases and 1.38 million deaths were reported due to this disease. 1 According to histological characteristics, this type of cancer can be divided into two main groups: small cell lung cancer, which comprises approximately 20% of cases; and non-small cell lung cancer, the vast majority of cases (approximately 80%). 2 Although progress has been made toward understanding the cellular and molecular biology of cancer and developing new treatments for this disease, the cumulative average survival rate for lung cancer remains very low. Five-year survival rates can vary from 13 to 20% in developed countries to 12% in developing countries. 3 Therefore, there is a pressing need for the development of new therapeutic strategies that are more effective and less toxic.
Nature is a rich source of inspiration for the development of novel drugs, and it is estimated that approximately 60% of the drugs used in cancer therapy are derived from natural sources or obtained through structural modification and the synthesis of natural-based compounds. 4 (R)-Goniothalamin (R-GNT) is a styryl lactone from plants of genus Goniothalamus Hk. f. et Thoms. (Family Annonaceae AL de Jussieu 1789 nom. conserv., the Custard Apple Family). 5 It is a natural product that has attracted the attention of the scientific community due to its high potential to induce cell death by apoptosis.
Its significant cytotoxicity in malignant cells and low cytotoxicity in normal cells are the properties that have produced great interest on this secondary metabolite. 6,7 Previous studies have demonstrated that R-GNT exhibits antiproliferative activity against breast, 7 –12 hepatoma, 8 leukemia, 13 –15 colon, 7,9 lung, 7,9 kidney, 9,16 ovary, and prostate 9 tumor cell lines. Modulation of apoptosis is one of the mechanisms of primary interest for the development of new therapeutic strategies for the cancer treatment. Studies have demonstrated that the antiproliferative activity of R-GNT involves the participation of this process. 8,10 –12 , 16,17
(S)-Goniothalamin (S-GNT; +-(6S)-5,6-dihydro-6-styryl-2-pyrone; Figure 1) is a synthetic enantiomer of R-GNT. Enantiomers are mirror images nonsuperposable that may have different biological activities. S-GNT has showed an even more significant antiproliferative activity in some cell lines. 18 Although its mechanism of action is largely unknown, a previous study has shown several differences in relation to the natural enantiomer, R-GNT. In renal cancer 786-O cells exposed to R-GNT and S-GNT, it was noted that R-GNT induced cell death by apoptosis, whereas S-GNT induced cell death by autophagy and apoptosis. 16
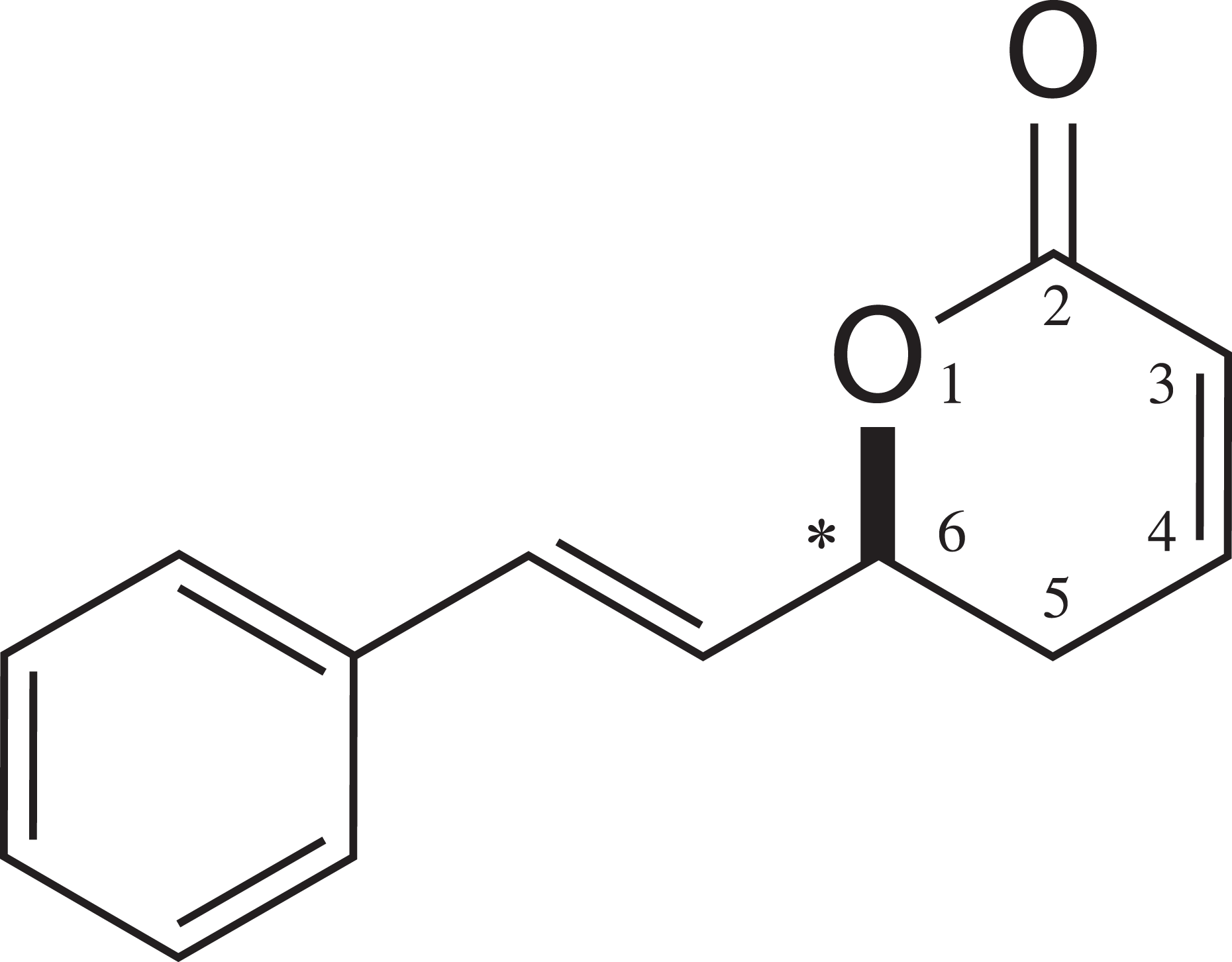
Chemical structure of (S)-goniothalamin; *indicates chiral center.
Apoptotic pathway alterations represent one of the hallmarks in tumor development 19 and may confer resistance to chemotherapy and radiotherapy, which contributes to neoplastic cell expansion. In lung cancer, some of the key changes found in regulating this process occur in the B-cell lymphoma 2-associated X protein (BAX) and p53 tumor protein (TP53) genes and in inhibitor of apoptosis protein (IAP) family members. 20
The proapoptotic Bax protein, which is encoded by BAX gene , is the most studied protein among the members of the Bcl-2 family. It is found as a 21 kDa monomer that is preferentially localized in the cytoplasm. In response to an apoptotic stimulus, Bax is transported to the outer mitochondrial membrane, and it allows extravasations of effector molecules to the cytosol contributing to the execution of apoptosis. 21
The p53 protein, encoded by TP53 gene, is a transcription factor considered as the “guardian of the genome.” This gene is able to regulate the expression of several other genes that have a variety of functions including inhibition of the cell cycle and apoptosis. This protein is involved in several stress response pathways that inhibit the growth and survival of potentially malignant cells. Under normal cellular conditions, p53 protein levels are kept very low, and during cellular stress situations such as oncogenic activation, genotoxic damage, lack of nutrients, hypoxia and others, the cellular levels of this protein increase. According to the cellular environment, with an increase in the cellular p53 level, the transcriptional transactivation of target genes such as BAX, IAPs, and several others occurs. 22
IAP is a family of regulatory proteins that play important roles in the cell such as inhibition of apoptosis and cell cycle progression. In humans, eight proteins belonging to this family have been identified: XIAP, HIAP1, HIAP2, survivin, NAIP, livin, Ts-IAP, and apollon. 23 Among them, survivin is encoded by the baculoviral IAP repeat-containing 5 (BIRC5) gene, and it is highly expressed in cancer cells but rarely expressed in differentiated normal tissues. Its increased expression in patients with cancer is an unfavorable prognostic marker correlated with decreased overall survival in various malignancies, including non-small cell lung cancer. Additionally, the ability of this IAP to inhibit apoptosis and regulate the cell cycle makes it an attractive target for the development of new therapeutic strategies for the treatment of malignant neoplasms. 24
S-GNT is an important target of study because there are only few studies in the literature, and its mechanism of action remains unknown. In this work, we investigated the cytotoxicity and cell proliferation kinetics, induction of apoptosis in situ, genotoxicity and expression of genes related to cell cycle, and apoptotic process including BAX, TP53, and BIRC5 after the exposure of non-small cell lung cancer (NCI-H460) cells to S-GNT. The choice of the NCI-H460 cells was based on relevance in the study of new therapeutic strategies for this type of cancer, which is one of the most common forms with the highest mortality rates in the world.
Material and methods
Chemical agents
The synthesis of S-GNT was performed according to the procedure of de Fátima et al. 18 in collaboration with Prof. Ângelo de Fátima, Departamento de Química, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil. The test substance was originally dissolved in dimethyl sulfoxide (DMSO) obtained from Mallinckrodt Chemicals (St. Luois, MO, USA) at a concentration of 0.05 M. The stock solution was diluted to 500 μM in Dulbecco’s Modified Eagle Medium (DMEM; Gibco®, Life Technologies, Carlsbad, CA, USA) and stored at −20°C. Before use, the 500-μM solution was diluted to the tested concentrations of 2.5, 12.5, and 25 μM. The dose selection was based on the study by de Fátima et al. 18 The DMSO concentrations did not exceed 0.05% (v/v) in culture.
The damage-inducing agents used in the experiments were doxorubicin (CAS 25316-40-9; Adriblastina®, Pharmacia, Italy) in the cytotoxicity test (18.4 μM), the cell proliferation kinetics (18.4 μM), and the comet assay (0.4 μM) and camptothecin (2.9 μM; CAS 7689-03-4; Acros Organics, Fisher Scientific Latin America Headquarters, Suwanee, GA, USA) for the assessment of apoptosis induction in situ. The dose selection of these agents was based on preliminary experiments.
Cell culture
The NCI-H460 cell line used was generously provided by Prof. JE de Carvalho, Universidade Estadual de Campinas, UNICAMP, Brazil. The cells were grown at 37°C and 5% carbon dioxide in DMEM supplemented with 10% fetal bovine serum (Gibco®) and antibiotics/antimycotics solution (Gibco®). Under these conditions, the cell cycle is 24 h. This cell line expresses p53 messenger RNA (mRNA) levels comparable to those observed in normal lung tissue cells. 25
Cytotoxicity test—MTT assay
Cytotoxicity was evaluated using the MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide; Invitrogen, Life Technologies, USA) described by Mosmann
26
with modifications. Briefly, cells were seeded in 24-well plates at a density of 2 × 104 cells/well in 500 μL medium per well and grown for 24 h for stabilization. After this period, the medium was removed and the cells were incubated in the same volume of fresh medium-containing S-GNT at a final concentration of 2.5, 12.5, and 25 μM for 24 and 48 h. After each experimental time, the medium was removed and the cells were incubated with MTT solution (500 μL of 0.1 mg/mL solution per well) at 37°C for 4 h. Next, the MTT solution was removed, and the formazan product crystals were dissolved in 500 μL DMSO. The absorbance at 540 nm was measured using a spectrophotometer (Uniscience, São Paulo, SP, Brazil). The assay was performed in three independent experiments and four replicates. Cell viability was expressed as the percentage relative to control group in absorbance:

Cell proliferation kinetics
The cell proliferation kinetics was conducted according to the study by Mauro et al. 27 Approximately 105 cells were seeded in culture tubes (10 cm2) and exposed to S-GNT at the concentrations of 2.5, 12.5, and 25 μM. After 24, 48, and 72 h, the cells were trypsinized (0.1% trypsin–ethylenediaminetetraacetic acid (EDTA), 37°C). The cell suspension was then centrifuged (5 min, at 84g), resuspended in 0.5 mL culture medium, and subsequently was transferred to a Neubauer chamber for cell counting. The assay was performed in two independent experiments.
Assessment of apoptosis induction in situ
Identification of apoptotic cells for morphological analysis was performed after staining with acridine orange according to the protocol of Rovozzo and Burke 28 with modifications from Tsuboy et al. 29 NCI-H460 cells were seeded into six-well plates (with cover slips) at an initial density of 1.2 × 105 cells/well. After 24 h of stabilization, the cells were exposed at 2.5, 12.5, and 25 μM S-GNT for 24 h. Then, the cover slips were removed, and the cells were fixed with Carnoy’s solution (methanol:glacial acetic acid, 3:1 v/v) for 5 min. Next, the cover slip was quickly incubated in decreasing concentrations of ethanol (from 95% to 25%). These incubations were followed by washing for 5 min with McIlvaine buffer (0.1 M citric acid and 0.2 M disodium phosphate), staining with acridine orange (0.01%) for 5 min, and a second wash with the buffer. In each group, 500 cells were analyzed via fluorescence microscopy (filter excitation of 420–490 nm and barrier filter 520 nm; Nikon, model 027012, Melville, NY, USA). The assay was conducted in three independent experiments. Cells that displayed condensation of chromatin and nuclear fragmentation were considered to be apoptotic.
Comet assay
The comet assay was performed in accordance with the assumptions of Tice et al. 30 Approximately 106 cells were seeded in culture flasks (25 cm2) and incubated for 24 h for stabilization. After the stabilization period, the cells were exposed at 2.5, 12.5, and 25 μM S-GNT for 3 h. After the experimental time, the cells were trypsinized (500 μL of 0.1% trypsin–EDTA, 37°C); the cell suspension was centrifuged (5 min, at 84g) and resuspended in 500 μL of DMEM. Subsequently, 20 μL of the cell suspension was mixed with 120 μL of low-melting point agarose (0.5%), deposited directly onto a slide pregelatinized with agarose (normal melting point, 1.5%), and then covered with a cover slip. After solidification of the agarose gel, the cover slip was removed; the slides were placed in a lysis buffer (2.5 M sodium chloride, 100 mM EDTA, 10 mM tris(hydroxymethyl)aminomethane (Tris), pH 10; 1% Triton X-100, and 10% DMSO) for approximately 1 h at 4°C. Slides were placed in an alkaline electrophoresis buffer solution (200 mM EDTA, 10 N sodium hydroxide, and pH > 13) for 20 min for DNA unwinding before electrophoresis (300 mA, 25 V, 20 min). Subsequently, the slides were neutralized (0.4 M Tris and pH 7.5) for 20 min and stained with 100 μL of 0.002 mg/mL ethidium bromide solution.
The analysis was conducted using fluorescence microscope (filter excitation of 420–490 nm and barrier filter 520 nm). Determination of damage was carried out by visual scoring and classification into four categories based on tail length: Class 0, undamaged cells showing no tail; Class 1, cells with a tail size less than the diameter of the nucleoid; Class 2, cells with a tail size two times longer than the diameter of nucleoid; Class 3, cells with a tail size greater than twice the diameter of the nucleoid. In total, 100 cells were analyzed per slide, and three independent experiments were conducted. Damage index (DI) was based on the length of DNA migration and calculated as follows:
Real-time reverse transcriptase quantitative polymerase chain reaction
The real-time reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) was conducted according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines. 31 Approximately 106 cells were seeded in culture flasks (25 cm2) and incubated for 24 h for stabilization. After the incubation period, samples (control; 25 μM S-GNT) were made, and after 12 h of treatment, total RNA was isolated using the Trizol® LS reagent (Invitrogen, Life Technologies, USA), according to the manufacturer’s protocol. RNA integrity was confirmed through electrophoresis on an agarose gel (1%), and the purity was determined on the spectrophotometric values at the A 260/A 280 absorbance (BioPhotometer, Eppendorf AG, Hamburg, Germany). The synthesis of complementary DNA (cDNA) was performed using 1 μg of total RNA, 0.5 pM oligo dT primer, 0.25 mM deoxyribonucleotide triphosphate, 40 U RNAseOUT™ Recombinant Ribonuclease Inhibitor, and 200 U M-MLV reverse transcriptase (1 μL; Invitrogen, Life Technologies, USA).
Real-time PCR was performed in a PTC 200 DNA Engine Cycler using the Chromo4 detection system (MJ Research, Bio-Rad, Waltham, MA, USA). Amplifications were performed in a final volume of 20 μL-containing 10 μL Platinum SYBR Green qPCR Supermix-UDG (Invitrogen, Life Technologies, USA), 0.375 μM of every primer (forward and reverse), and 2 μL template. The PCR conditions were as follows: an initial step at 50°C for 1 min; 95°C for 3 min; 35 cycles of 95°C for 20s, 60°C for 30s, and 72°C for 20s; 95°C for 10s; and 40°C for 1 min. The melting curve analysis was carried out at the end of the reaction with temperature range from 50°C to 95°C every 0.5°C for 5s. The data were normalized with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene. Primers for transcripts amplification of TP53, BIRC5, and BAX are showed in Table 1. All experiments were conducted with two independent cultures, and each cDNA sample was analyzed with three mechanical repetitions for each primer pair.
Sequence of primers used in RT-qPCR.

BIRC5: baculoviral inhibitor of apoptosis repeat-containing 5; TP53: p53 tumor protein; BAX: B-cell lymphoma 2-associated X protein; RT-qPCR: reverse transcription quantitative polymerase chain reaction; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; F: forward transcription; R: reverse transcription.
Statistical analyses
Data obtained from MTT assay, cell proliferation kinetics, and comet assay were subjected to analysis of variance, followed by Dunnett’s test (p < 0.05). The assessment of apoptosis induction in situ data was subjected to Fisher’s exact test. All statistical tests were conducted using GraphPad Prism® version 5.00 software (GraphPad Software, San Diego, California, USA.) In the study of gene expression by RT-qPCR, analyses of the relative values of gene expression and normalization by the GAPDH gene were performed as described by Pfaffl et al., 35 using the Relative Expression Software Tool-384 (REST-384 software, Sidney, Australia).
Results
S-GNT induces cytotoxicity and reduces cell growth
To estimate the effects of S-GNT on cell viability of NCI-H460 cells, MTT assay was performed. Our results showed that S-GNT reduced cell viability in a concentration-dependent manner. After 24 h of exposure, 25 μM S-GNT decreased the viability of NCI-H460 cells to 89.8%. After 48 h, all tested concentrations (2.5, 12.5, and 25 μM) decreased the cell viability. The minimum cell viability observed was 74.7% when NCI-H460 cells were exposed to 25 μM S-GNT for 48 h (Figure 2)

Cell viability percentage of NCI-H460 cells exposed to S-GNT for 24 and 48 h calculated from the absorbance values obtained from the MTT assay. The bars represent the mean ± SD obtained in three independent experiments. Statistical differences: *p < 0.05 and **p < 0.01 compared with the control. NCI-H460: non-small cell lung cancer cell; S-GNT: (S)-goniothalamin; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide.
Furthermore, we investigated the effects of S-GNT on the cell proliferation kinetics through a cell growth curve. S-GNT showed a decrease in cell proliferation in a concentration-dependent manner. The inhibition of cell growth was noted when NCI-H460 cells were exposed to 12.5 and 25 μM S-GNT for 48 and 72 h (Figure 3).

Cell growth curve determined by cell count after 24, 48, and 72 h of NCI-H460 cells exposure to S-GNT. Statistical difference: *p < 0.05 and **p < 0.01 compared with the control. NCI-H460: non-small cell lung cancer cell; S-GNT: (S)-goniothalamin.
The cytotoxic-inducing agent (18.4 μM doxorubicin) showed cytotoxicity after 24 and 48 h of exposure. It also reduced the cell growth at 48 and 72 h.
S-GNT induces apoptosis
To assess the effect of S-GNT on apoptosis induction, we conducted acridine orange staining to observe morphological changes in nucleus. Our results showed an increase in cells with morphological changes, including condensation of chromatin and nuclear fragmentation in NCI-H460 cells exposed to 25 μM S-GNT for 24 h (Table 2). However, S-GNT was not so sensible to apoptosis induction, and the percentage of apoptotic cells observed was 5.2%.
Assessment of apoptosis induction in situ after 24 h of NCI-H460 cells exposure to S-GNT.a
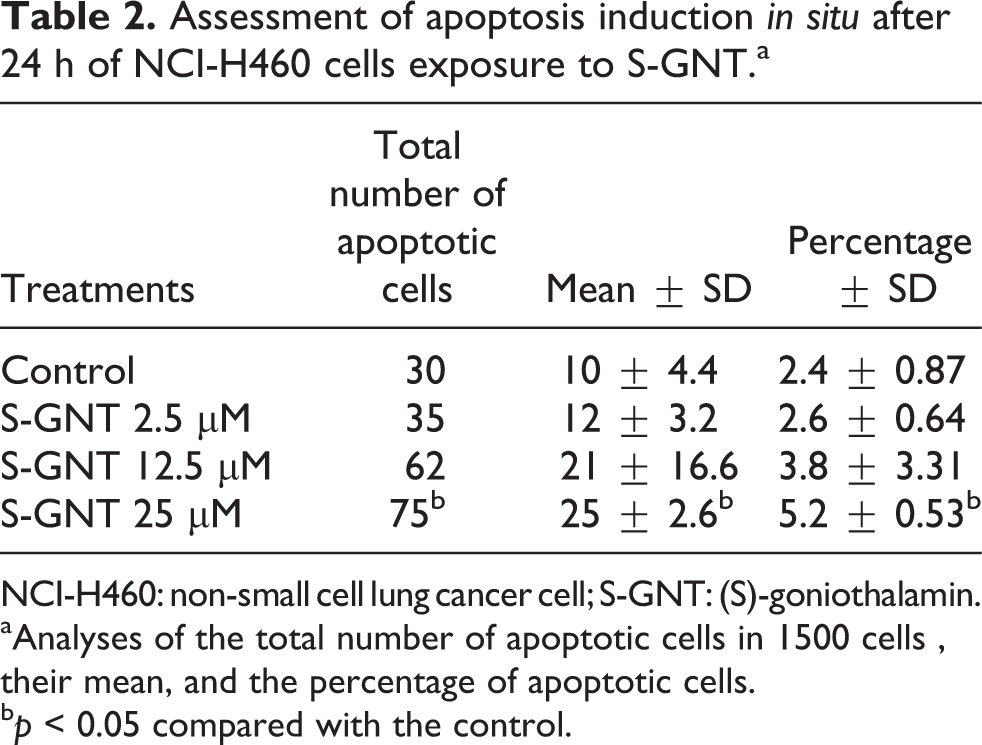
NCI-H460: non-small cell lung cancer cell; S-GNT: (S)-goniothalamin.
aAnalyses of the total number of apoptotic cells in 1500 cells , their mean, and the percentage of apoptotic cells.
b p < 0.05 compared with the control.
Exposure to the apoptosis-inducing agent (2.9 μM camptothecin) led to an increase in the apoptosis induction.
S-GNT induces DNA damage
The comet assay was conducted to examine whether S-GNT induces DNA damage in NCI-H460 cells. Our results showed that S-GNT led to an increase in the length of DNA migration to tail at all evaluated concentrations, as indicated by DI (Figure 4). The DI observed was 57 for NCI-H460 cells exposed to 2.5 μM S-GNT, 89 for 12.5 μM S-GNT, and 93 for 25 μM S-GNT. Thus, there was higher frequency of cell damage when the concentration of S-GNT increases, suggesting that the induced genotoxicity occurred in a concentration-dependent manner (Table 3). DNA damage-inducing agent (0.4 μM doxorubicin) treatment led to an increase in DI.
Mean number of positive cells in comet assay, distribution among the different classes of DNA damage, and DI obtained in control and S-GNT treatments, after 3 h of exposure.a

DOX: doxorubicin; DI: damage index; S-GNT: (S)-goniothalamin.
aControl; DOX: 0.4 μM; 2.5, 12.5, and 25 µM S-GNT concentrations.
b p < 0.01 compared with the control.
c p < 0.05 compared with the control.
d p < 0.001 compared with the control.
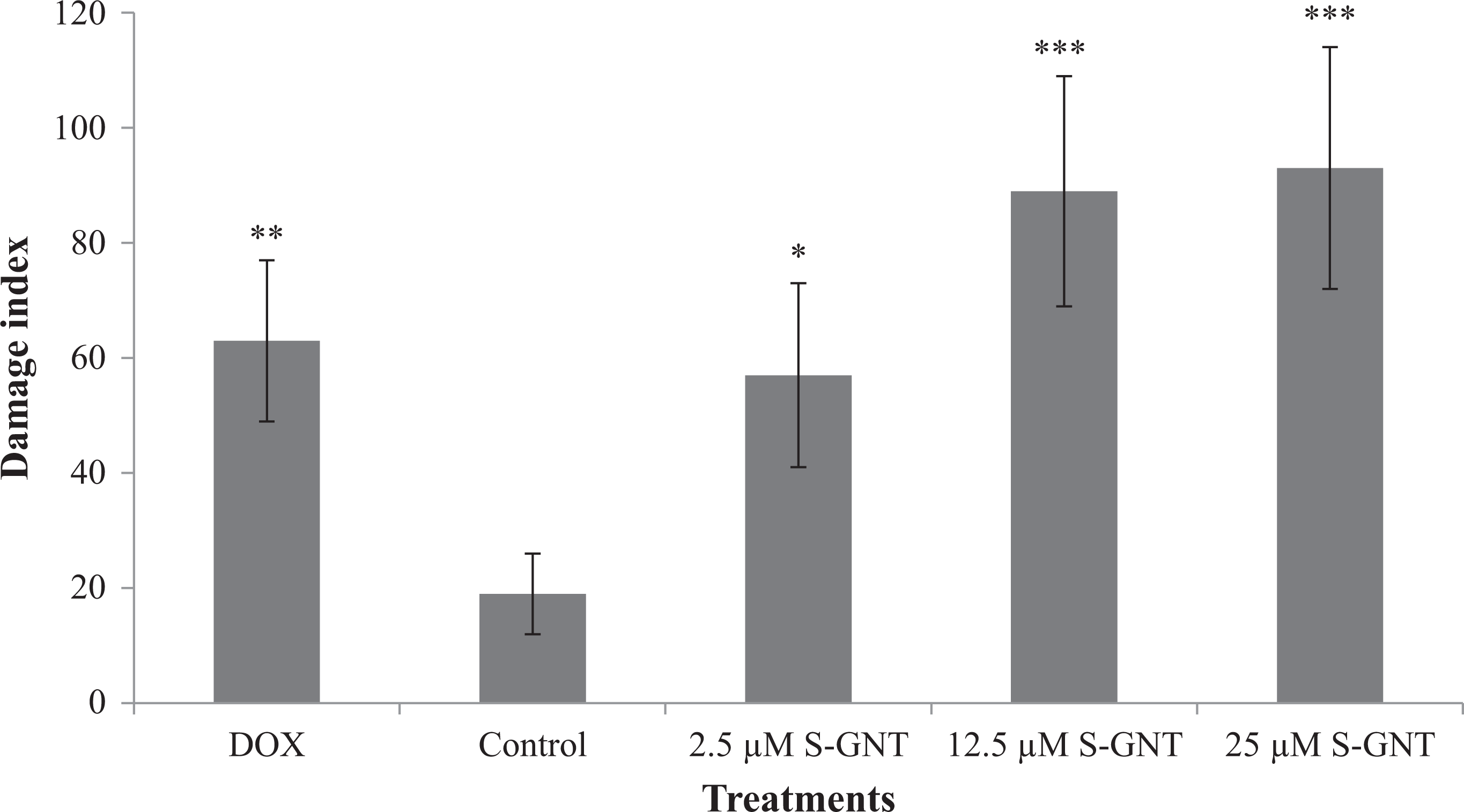
DI observed after exposure of NCI-H460 cells to S-GNT for 3 h. DOX: 0.4 μM doxorubicin; control; and 2.5, 12.5, and 25 µM S-GNT concentrations. The bars represent the mean ± SD obtained in three independent experiments. Statistical difference: *p < 0.05), ** p < 0.01) and ***p < 0.001) compared with the control. DI: damage index; NCI-H460: non-small cell lung cancer cell; S-GNT: (S)-goniothalamin.
The cell viability analysis using the trypan blue stain exclusion method showed an index greater than 80% in all the samples.
S-GNT downregulates BIRC5 gene expression
mRNA levels of BIRC5, TP53, and BAX genes were analyzed using RT-qPCR. Our results showed downregulation in BIRC5 mRNA levels (1.697-fold change) in NCI-H460 cells exposed to 25 μM S-GNT for 12 h compared with the control group (p = 0.001). There was no change in mRNA levels of TP53 and BAX genes (Figure 5).

Relative expression of the BIRC5, TP53, and BAX genes using RT-qPCR after 12 h of exposure to 25 μM S-GNT. Statistical difference: ***p = 0.001 compared with the control. BIRC5: baculoviral inhibitor of apoptosis repeat-containing 5; TP53: p53 tumor protein; BAX: B-cell lymphoma 2-associated X protein; RT-qPCR: reverse transcription quantitative polymerase chain reaction.
Discussion
Several studies have indicated GNT as a potent antiproliferative agent because of its potential to induce apoptosis in tumor cells. However, few studies have examined the activity of the synthetic enantiomer S-GNT. Therefore, the goal of this work was to study the effects of this molecule in NCI-H460 cell line. The results demonstrated that S-GNT reduces cell viability in a dose-dependent manner, induces apoptosis, demonstrates genotoxic effects, and reduces the BIRC5 mRNA levels.
The cytotoxic potential of R-GNT against various tumor cell lines has been reported. 6 –16 Studies showed that GNT is a good cytotoxic compound against large cell lung carcinoma COR-L23 (half maximal inhibitory concentration (IC50): 17.55 μM) 7 and also to NCI-H460 cells (IC50: 6.4 μM). 9 Our results suggest that the synthetic enantiomer also displays this capability, as indicated by cell viability reduction in NCI-H460 cells after S-GNT exposure. In agreement with this result, changes in the cell proliferation kinetics were also noted. In 2006, de Fátima et al. 18 also demonstrated dose-dependent cytotoxic activity of S-GNT against various tumor cell lines exposed to concentrations between 0.25 and 250 μg/mL.
The cytotoxic effect observed using the MTT assay may have occurred due to the inhibition of cell growth and may be a result of the inhibition of mitochondrial metabolism and/or the induction of apoptosis. Cell death by apoptosis is a process of great interest in the development of therapies for the cancer treatment. Various authors have suggested that this process is the main mechanism by which R-GNT plays its antiproliferative role. 8,10 –13,16,17,36
As with R-GNT, we observed that S-GNT demonstrated the potential to induce apoptosis in NCI-H460 cells in a concentration-dependent manner. In this study, it was noted that S-GNT at a dose of 25 μM induced 5.2% of cell death via apoptosis after 24 h of exposure. These results suggest that the induction of apoptosis is directly related to the cytotoxic potential of this compound. This result is in accordance with the work of de Fátima et al., 16 which demonstrated the ability of S-GNT to induce apoptosis in 786-O kidney cancer cells. In this study, there was an increase in the cleavage of poly adenosine diphosphate–ribose polymerase, an indicator of the apoptotic effect after exposure at 4 nM S-GNT (IC50 value for 786-O cells).
The mechanisms by which GNT induces cell death are not completely elucidated, but several studies have suggested that the main pathway leading to cell apoptosis is intrinsic or mitochondrial pathway. Lee et al. 11 demonstrated that after exposure of MCF-7 cells to styryl pyrone derivative, a compound extracted from Goniothalamus umbrosus, the intrinsic pathway of apoptosis was activated. The authors observed the accumulation of cytochrome c and activation of initiator caspase-9 and effector caspase-7. In 2003, Inayat-Hussain et al. 13 also showed that GNT induces apoptosis in human promyelocytic leukemia (HL-60) cells by activation of intrinsic pathway, with loss of mitochondrial membrane potential, and the activation of initiator caspase-9 and effector caspase-3 and -7. Chan et al. 17 reported an increase in the activities of caspase-2, -3, and -9 without the activation of cas-pase-8, which characteristically is activated via death receptors (i.e. the extrinsic pathway), after exposure of coronary artery smooth muscle cells (CASMCs) to GNT.
The intrinsic pathway is activated in response to death stimuli occurring within a cell, such as DNA damage. Several studies have shown that R-GNT displays genotoxic effects. These studies suggest that these effects may be the main mechanism by which R-GNT triggers apoptosis because when DNA damage is not repaired, the apoptotic mechanism is triggered. 37 Chan et al. 36 showed concentration-dependent genotoxicity using comet assay in vascular smooth muscle cells exposed to GNT (0.5 and 1.5 μg/mL). These authors have suggested that the induction of DNA damage is responsible for the cytotoxicity caused by this compound, mainly through the mechanism of apoptosis. Inayat-Hussain et al. 38 showed which DNA damage induction after exposure of Jurkat leukemia T cells to 50 μM GNT resulted in activation of the intrinsic pathway of apoptosis. Rajab et al. 14 found that the treatment of leukemic cell lines HL-60 and Human T4 lymphoblastoid cell line CEMSS with GNT at a concentration equivalent to the 10% inhibitory concentration (IC10) and IC25 (IC50s of 4.5 and 2.4 μg/mL, respectively) resulted in the induction of DNA damage as detected by the comet assay. Umar-Tsafe et al. 39 also observed a significant increase in genotoxicity in Chinese hamster ovary cells after exposure to 5 and 10 μM GNT as determined by a chromosomal aberration assay. Additionally, the analysis of DNA damage before and after metabolic activation demonstrated that this molecule acts as a direct mutagen, because damage is induced with or without metabolic activation. Some studies suggest that the capacity of GNT to induce damage to genetic material is directly related to the occurrence of oxidative stress. 12,36
In this study, the analysis of genotoxicity using the comet assay has shown that S-GNT induces DNA damage in a concentration-dependent manner. In NCI-H460 cells, although the induction of DNA damage has been detected in all used concentrations, only the highest concentration (25 μM) significantly induced apoptosis, suggesting that the mechanism of repair has recovered part of the cells that have suffered DNA damage.
Although the apoptotic mechanism may be independent of p53 protein, apoptosis as a consequence of DNA damage often occurs in a p53-dependent manner, which modulates the susceptibility of cells to apoptosis by activating proapoptotic genes such as BAX. 40 In this study, no significant changes in the mRNA levels of TP53 and BAX were observed even with the induction of damage to genetic material caused by S-GNT. This result differs from that of de Fátima et al., 16 where a reduction in expression of the proapoptotic Bax protein was observed after exposure of 786-O cells to S-GNT. This result also differs from the work of Chan et al., 17 where an increase in the p53 protein levels was detected after exposure of CASMCs to R-GNT.
An important result related to the mechanism of action of S-GNT was showed for the first time in this study and may contribute to explain the process of inhibition of cell proliferation and the induction of apoptosis in tumor cells caused by this compound. This result was the significant reduction in BIRC5 gene expression. This gene encodes the protein survivin, which has two important functions in the cell. It can regulate the cell cycle 41,42 and suppress apoptosis. 43 –45
Survivin plays an important role in cell cycle regulation. During mitosis, survivin associates with polymerized tubulin, centrosome, metaphase microtubules, and the anaphase zone. 41 Cell division in the absence of survivin results in defects in chromosome alignment, failed cytokinesis, and eventual cell death, which in this case is related to aberrant cell division. 42
With regard to apoptosis, the idea that survivin demonstrates inhibitory potential is well established and has been demonstrated in various studies, indicating that its overexpression results in the inhibition of apoptosis 43 –45 and its inhibition by antagonists leads to a caspase-dependent mode of cell death. 46 Furthermore, there are studies indicating that its inhibition increases the cytotoxicity of chemotherapeutic agents by reverse chemoresistance of tumor cells to these drugs, indicating that this protein is an important survival factor in tumors. 47 Thus, drugs that negatively regulate the expression of survivin, such as S-GNT, are important for the cancer treatment.
Several authors have reported an increase in the activation of caspases-3, -7, and -9 after treatment with GNT. 5,10,11 Survivin has antiapoptotic potential due to its capacity to bind to SMAC/DIABLO factor and also interacting with XIAP favoring caspases inhibition. Therefore, in accordance with our data, the downregulation of BIRC5 gene after exposure to S-GNT may be one of the mechanisms responsible for the apoptosis induction.
Conclusions
This study demonstrated that the S-GNT has the potential to induce cell death by apoptosis, possibly by inducing DNA damage and by the downregulation of BIRC5 gene without changing the mRNA levels of the TP53 and BAX genes. This molecule thus presents itself as a potential therapeutic agent for lung cancer treatment. However, further studies are necessary to discover additional information regarding the mechanism of action, mainly concerning its ability to regulate the expression of BIRC5 gene.
Footnotes
Conflict of interest
The authors declared no conflicts of interest.
Funding
This work was supported by CNPq, CAPES, and the Fundação Araucária.