
Abstract
Humans are commonly exposed to nicotine, one of the most important lifestyle chemicals. The occurrence of high levels of arsenic in the groundwater of the southeast region of Asia has received much attention in the past decade and has become a global health concern. Predominant occurrence of both these chemicals and ease of their human exposure led us to investigate the effect of nicotine, a major tobacco alkaloid, on arsenic toxicity. Adult male rats were pre-exposed to two different doses of nicotine (0.75 and 3 mg/kg, intraperitoneally) for 7 days followed by 30 days of arsenic exposure (50 ppm sodium arsenite in drinking water). Nicotine pre-exposure resulted in an increased brain arsenic accumulation and a decreased liver arsenic concentration. Arsenic also caused a significant oxidative stress in the blood, brain and liver of the exposed rats. Glutathione-S-transferase, a phase II enzyme, was inhibited by both arsenic and nicotine but no such inhibition was noted in arsenic-treated animals pre-exposed to nicotine. Upon nicotine pre-exposure, brain acetylcholinesterase increased, while monoamine oxidase (MAO) decreased. The toxic effects of MAO significantly attenuated with nicotine pre-exposure. The present study suggests that nicotine may not be the major contributing factor for the previously reported synergistic toxic interaction between tobacco and arsenic. Nicotine pre-exposure in arsenic-exposed animals revealed interesting toxicokinetics and oxidative stress modulating interactions in the brain and liver of rats, which requires further exploration.
Introduction
Arsenic is one of the most widely studied metalloid. High levels of arsenic exist in the nature as inorganic arsenic (iAs) as well as organic forms. 1 –3 Humans might get exposed to arsenic via air and food but it has been reported that in many developing countries, the major route of exposure to iAs is through contaminated drinking water. 4 Arsenicosis, a pathological condition following chronic arsenic exposure, is reported to affect more than 200 million people worldwide with approximately 38 million residing only in the Indo-Bangladesh region. 5 –7 Human exposure to iAs may result in degenerative, inflammatory and neoplastic manifestations of skin, respiratory system, blood, lymphatic system, nervous system and reproductive system. 8 –10 Although the mechanism for arsenic toxicity can be multifactorial, oxidative stress is one of the most widely studied and accepted mechanisms. 11 Nicotine is a natural, water-soluble alkaloid present in the leaves of tobacco, Nicotiana tobacum, and Coca. Nicotine, following inhalation during smoking, gets rapidly absorbed into circulatory system and distributed with more than 80% being metabolized in the liver. 12 The main sources of nicotine exposure to humans are tobacco products. Tobacco smoking is expected to be the single biggest cause of mortality worldwide by 2030, killing about 10 million people per year. 13 Nicotine poisoning causes nausea, vomiting, abdominal pain, diarrhoea, headache and sweating, whereas more severe poisoning may result in dizziness, weakness, confusion, hypotension and coma. 14 Although nicotine contributes to tobacco-related adverse health effects, yet its specific health effects have not been confirmed since it is taken simultaneously with other harmful substances, present in tobacco smoke.
Thus, arsenic and nicotine are of great interest to the researchers due to their direct and close impact on human health. Although the two chemicals have been studied individually in animal models and human subjects, their direct interaction has never been investigated. Few recent reports have investigated and indicated possible interactions between tobacco and arsenic. 15,16 These reports have been reviewed in the present study since nicotine forms the major active constituent of tobacco. Clinical evidence also suggests that concurrent exposure to arsenic and tobacco smoking synergistically increases the risk of lung and bladder cancer. 17 –21 These findings have been supported by animal experiments since arsenic and cigarette smoke synergistically caused DNA damage. 16 Furthermore, arsenic metabolism may be a risk-modifying factor for arsenic-related health effects in the presence of tobacco. The interactions were suggested to be due to tobacco smoking-induced immunosuppression, which may lead to the inhibition of arsenic methylation. 22 Cigarette smoke and arsenic co-exposure were also been reported to induce lung injury via oxidative stress. 23 –25 Both the toxicants promote reduced glutathione (GSH) depletion with consequent effects on proteins, lipids and DNA oxidation. 16 Thus, despite being an area of utmost environmental health concern, only few studies investigated the interaction between tobacco and arsenic. Thus, the previously reported data supporting synergism between tobacco and arsenic are possibly mediated through oxidative stress or by altered arsenic metabolism. Based on our review of the previous literature, the present study was aimed at evaluating (i) whether any toxicological interactions occur between nicotine, as the major constituent of tobacco, and arsenic, (ii) the role of oxidative stress in the possible nicotine–arsenic interaction and (iii) the effect of nicotine on arsenic concentration in the target organs. The present study thus, for the first time, investigates whether the reported tobacco–arsenic toxic synergism is possibly due to its nicotine constituent.
Materials and methods
Chemicals and reagents
Sodium meta-arsenite was obtained from Merck (Darmstadt, Germany). Nicotine hydrogen tartarate was procured from Sigma (St Louis, Missouri, USA). All other laboratory chemicals and reagents were purchased from Merck (Darmstadt, Germany), Sigma or Acros Organics (New Jersey, USA) and were of analytical reagent or extra-pure grade.
Animals
A total of 30 male albino adult rats of Wistar strain weighing 120 ± 20 g were obtained from animal house facility of Defence Research and Development Establishment (DRDE), Gwalior. Animal ethical committee of DRDE, Gwalior, India, approved the experimental protocol (Approval number AH/05010/2010-11, dated 20 August 2011). Experimental animals were housed in stainless steel cages in an air-conditioned room with temperature maintained at 25 ± 2°C and 12 h alternating day and night cycles. Rats were fed with standard diet and water ad libitum throughout the study period.
Treatment regimen
Experimental animals were acclimatized for a week, prior to the beginning of the dosing regimen. During the experimental tenure, animals were weighed weekly and the dose volume adjusted accordingly with a maximum of 0.5 ml per rat, intraperitoneally (i.p.). Doses of nicotine and arsenic were selected based on our previous studies and preliminary data generated from pilot studies in our laboaratory. 26,27 Animals were pre-exposed and treated as below:
Group I – control
Group II – nicotine (6 mg/kg; i.p.)
Group III – arsenic in drinking water (50 ppm)
Group IV – nicotine (0.75 mg/kg; i.p.) + arsenic (50 ppm)
Group V – nicotine (3 mg/kg; i.p.) + arsenic (50 ppm)
Nicotine pretreatment lasted for 1 week, which was later discontinued during 4 weeks of arsenic exposure in drinking water. After completion of the above-mentioned exposures, blood was drawn through retro-orbital route and collected in heparinized vials at 4°C. The animals were then killed by cervical dislocation under anesthesia. Brain and liver were removed, washed with normal saline, blotted and snap frozen and stored at −80°C until the biochemical assays and metal estimations were carried out. Immediately before performing biochemical assays, the tissue homogenates were prepared using a Potter–Elvehjem homogenizer (model RQ-127 A, Remi, Mumbai) fitted with a Teflon pestle under cold conditions.
Arsenic level determination
Arsenic level in blood, brain and liver was measured after wet acid digestion using a Microwave Digestion System (Multiwave-3000, Anton Paar, Graz, Austria). Tissue samples were weighed (1 ml blood or 1 g brain or liver sample) and digested in nitric acid and the volume was made constant (5 ml) for all samples. Arsenic was estimated using an hydride vapour generation system fitted with an atomic absorption spectrophotometer (AAS, Perkin Elmer model Analyst 100, Norwalk, CT, USA). 3
Haematological variables measurement
White blood cells, red blood cells (RBC), haemoglobin, haematocrit, mean cell volume, mean cell haemoglobin, mean cell haemoglobin concentration and platelet counts were measured on a Sysmex Hematology Analyzer (model K4500).
Biochemical assays
The amount of reactive oxygen species (ROS) in blood, brain and liver was measured using 2′,7′-dichlorofluorescin diacetate (DCF-DA). The assay was performed as described by Socci et al. 28 Briefly, 5% RBC haemolysate was prepared and diluted to 1.5% with ice-cold 40 mM Tris–hydrochloric acid (HCl) buffer (pH 7.4) and further diluted to 0.25% with the same buffer and placed on ice. Tissue homogenates were diluted to 1% with ice-cold 40 mM Tris–HCl buffer (pH 7.4). The samples were divided into two equal fractions. In one fraction 40 μl of 1.25 mM DCF-DA in methanol was added for ROS estimation. Another fraction in which 40 μl of methanol was added served as a control for tissue/haemolysate autofluorescence. All the samples were incubated for 15 min in a water bath at 37°C. Fluorescence was determined at 488 nm excitation and 525 nm emission wavelength using a fluorescence plate reader (Perkin Elmer, LS-55, Maryland, USA). Results were expressed in fluorescent intensity unit.
Glutathione level in blood (GSH) was measured following the method described by Ellman 29 and modified by Jollow et al. 30 Whole blood of 0.2 ml was added to 1.8 ml of distilled water and incubated for 10 min at 37°C for complete haemolysis. After adding 3 ml of 4% sulphosalicylic acid, tubes were centrifuged at 2500g for 15 min. Supernatant of 0.2 ml was mixed with 0.4 ml of 10 mM solution of 5,5-dithiobis-(2-nitro benzoic acid) in the presence of 1 ml phosphate buffer (0.1 M, pH 7.4). The absorbance was recorded at 412 nm.
On the other hand, tissue GSH and oxidized glutathione (GSSG) levels were measured fluorometrically using the method of Hissin and Hilf. 31 In the assay, o-phthalaldehyde (OPT) reacts with GSH (not GSSG) to generate strong fluorescence, so that GSH can be specifically quantified. To measure GSSG specifically, a GSH quencher, N-ethylmaleimide (NEM), is added to specifically quench GSH, and the GSSG level can be specifically quantified. Briefly, 0.25 g of tissue sample was homogenized on ice with 3.75 ml of 0.1 M phosphate–0.005 M ethylenediaminetetraacetic acid (EDTA) buffer (pH 8.0) and 1 ml of 25% metaphosphoric acid that was used as a protein precipitant. The homogenate (4.7 ml) was centrifuged at 100,000g for 30 min at 4°C. For the GSH assay, 0.5 ml supernatant and 4.5 ml phosphate buffer (pH 8.0) were mixed. The final assay mixture (2.0 ml) contained 100 μl supernatant, 1.8 ml phosphate–EDTA buffer and 100 μl OPT. For the GSSG assay, 0.5 ml supernatant was incubated at room temperature with 200 μl of 0.04 mol/l NEM solution for 30 min. To this mixture, 4.3 ml of 0.1 mol/l sodium hydroxide (NaOH) was added. A 100-μl sample of this mixture was taken for the measurement of GSSG using the procedure described above for the GSH assay, except that 0.1 mol/l NaOH was used as the diluent instead of phosphate buffer. The fluorescence for both GSH and GSSG was measured at an excitation wavelength of (λ Ex) 350 nm and emission wavelength of (λ Em) 420 nm. The reduced GSH and oxidized GSSG were expressed as their ratio.
Tissue lipid peroxidation was measured by the method of Ohkawa et al. 32 and modified by Saxena and Flora. 33 Whole tissue homogenate was incubated with 8.1% sodium dodecyl sulphate (w/v) for 10 min followed by addition of 20% acetic acid (pH 3.5). The reaction mixture was further incubated with 0.6% thiobarbituric acid (TBA) (w/v) for 1 h in a boiling water bath. A pink colour chromogen, so formed, was extracted in butanol pyridine solution and read at 532 nm. Lipid peroxidation was calculated using molar extinction coefficient of the MDA–TBA complex (1.56 × 105 M−1 cm−1). The level of TBA reaction substances (TBARS) was expressed as nanomoles of TBARS per gram of tissue weight.
Glutathione-S-transferase (GST) activity was determined following the procedure of Habig et al. 34 Supernatant obtained after centrifuging tissue homogenate at 1500g for 10 min followed by 10,000g for 30 min at 4°C was used for the GST assay. The reaction mixture contained 0.02 ml of 1-chloro-2,4-dinitrobenzene, 2.9 ml of GSH and 30 µl of post-mitochondrial supernatant. Change in colour was monitored by recording absorbance (340 nm) at 30 s intervals for 30 min. The activity of enzyme was expressed as nanomoles of GSH–1-chloro-2,4 nitrobenzene (CDNB) conjugate formed per minute per gram of tissue weight, using molar extinction coefficient of the conjugate (9.6 × 106 M− 1 cm− 1).
Activity of acetylcholinesterase (AChE) in brain was determined according to the method of Ellman et al. 35 A 10% brain homogenate (w/v) was prepared in 0.25 M sucrose and the activity of AChE was measured using acetylthiocholine as a substrate. The readings were taken at 412 nm for 5 min at 1 min interval and calculated using molar extinction coefficient of 5-mercapto-2-nitrobenzoic acid (13.6 × 103 M−1 cm−1). AChE activity is expressed as nanomoles of product formed per minute per milligram of protein. Monoamine oxidase (MAO) activity was studied in brain mitochondrial fraction following the method of Wurtman and Axelrod. 36 Briefly, 1.0 ml of 0.2 M phosphate buffer (pH 7.2) and 0.8 ml distilled water were added to 100 μl of mitochondrial fraction. A 0.1-ml of benzylamine HCl (0.1 M, pH 7.2) was added to the experimental tubes. The tubes were incubated for 30 min at 37°C and the reaction was stopped by adding 1 ml of 10% perchloric acid. In controls, instead of mitochondrial fraction, an equal amount of phosphate buffer was added. After centrifugation, the supernatant was diluted with equal volume of distilled water and read at 250 nm. The enzyme activity was expressed as nanomoles of benzaldehyde formed per minute per milligram of protein. Total tissue protein was measured by the method of Lowry et al. 37
Statistical analysis
Data were expressed as mean ± SEM. Data comparisons were carried out by one-way analysis of variance followed by the Bonferroni’s post hoc test to compare mean between the different treatment groups using the Software GraphPad InStat (San Diego, USA). The difference with p < 0.05 was considered significant.
Results
Arsenic level
Arsenic concentration measured in blood, brain and liver revealed interesting information. Animals exposed to arsenic show increased arsenic concentration in blood, brain and liver compared with control animal receiving arsenic-free drinking water (Figure 1). Animals pre-exposed to nicotine (0.75 and 3 mg/kg) followed by arsenic exposure also showed more pronounced blood arsenic level compared with the group exposed to arsenic alone. Brain and liver arsenic levels in animals pre-exposed to nicotine (both the doses) showed a significant increase and decrease, respectively, compared with arsenic alone.
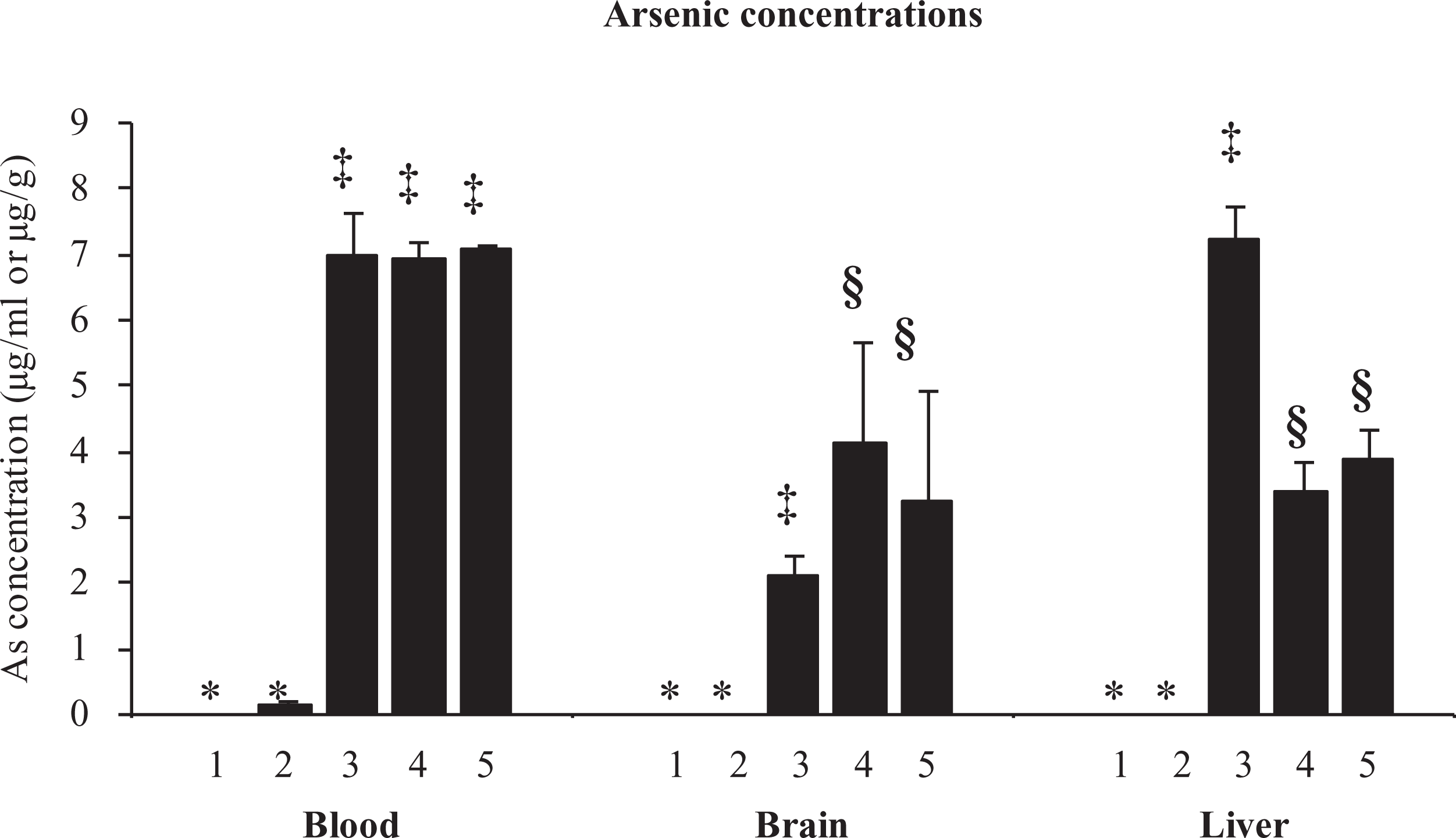
Effect of nicotine pre-exposure on arsenic level in blood, brain and liver of Wistar rats. Group I: control; group II:– nicotine; group III: arsenic; group IV: nicotine (0.75 mg/kg) + arsenic; group V: nicotine (3 mg/kg) + arsenic. Measured as micrograms per millilitre in blood and micrograms per gram tissue weight in brain and liver. Values are mean ± SEM; n = 5. *, ‡, §: differences between values with matching symbol notations within each column are not statistically significant at 5% level of probability.
Effect on haematological variables
No significant changes in any of the haematological variables following exposure to nicotine, arsenic or in combination were noted, except for an elevation in haemoglobin level on nicotine alone treatment. This effect however was not observed in animals pre-exposed to nicotine followed by arsenic (Table 1).
Effect of nicotine pre-exposure on haematological variables in arsenic exposed rats.a

RBC: red blood cells; WBC: white blood cells; Hb: haemoglobin; HCT: haematocrit; MCV: mean cell volume; MCH: mean cell haemoglobin; MCHC: mean cell haemoglobin concentration; PLT: platelet.
aValues are mean ± SEM; n = 5.
bDifferences between values with matching symbol notations within each column are not statistically significant at 5% level of probability.
cDifferences between values with matching symbol notations within each column are not statistically significant at 5% level of probability.
Effect on ROS
Animals exposed to arsenic alone showed significantly elevated blood ROS compared to controls (Figure 2). However, nicotine pretreated animals (both the doses) significantly reduced arsenic-induced elevated blood ROS levels. This trend was not observed in brain and liver of rats. Both nicotine and arsenic individually caused a significant increase in the ROS level; however, the level was further increased in brain, while decreased in liver in animals pretreated with nicotine (both the doses).

Effect of nicotine pre-exposure on arsenic-induced reactive oxygen species and TBARS in blood, brain and liver of Wistar rats. Groups: 1 – control, 2 – nicotine 3 – arsenic 4 – nicotine (0.75 mg/kg) + arsenic; group V: nicotine (3 mg/kg) + arsenic. ROS: reactive oxygen species as fluorescence intensity unit. TBARS expressed as nanomoles of TBARS per gram of tissue. Values are mean ± SEM; n = 5. *, ‡, §: differences between values with matching symbol notations within each column are not statistically significant at 5% level of probability. TBARS: thiobarbituric reactive substances.
Effect on TBARS
No significant change was observed in brain TBARS levels in any of the exposed groups. Liver TBARS significantly increased following exposure to arsenic or nicotine individually and nicotine (0.75 mg/kg) + arsenic co-exposed group compared with the control. On the other hand, pretreatment with high nicotine (3 mg/kg) followed by arsenic significantly decreased TBARS level compared with arsenic or nicotine alone groups (Figure 2).
Effect on GSH in blood
As shown in Figure 3, GSH level showed depletion in blood of animals receiving nicotine, arsenic alone or in combination compared with control.
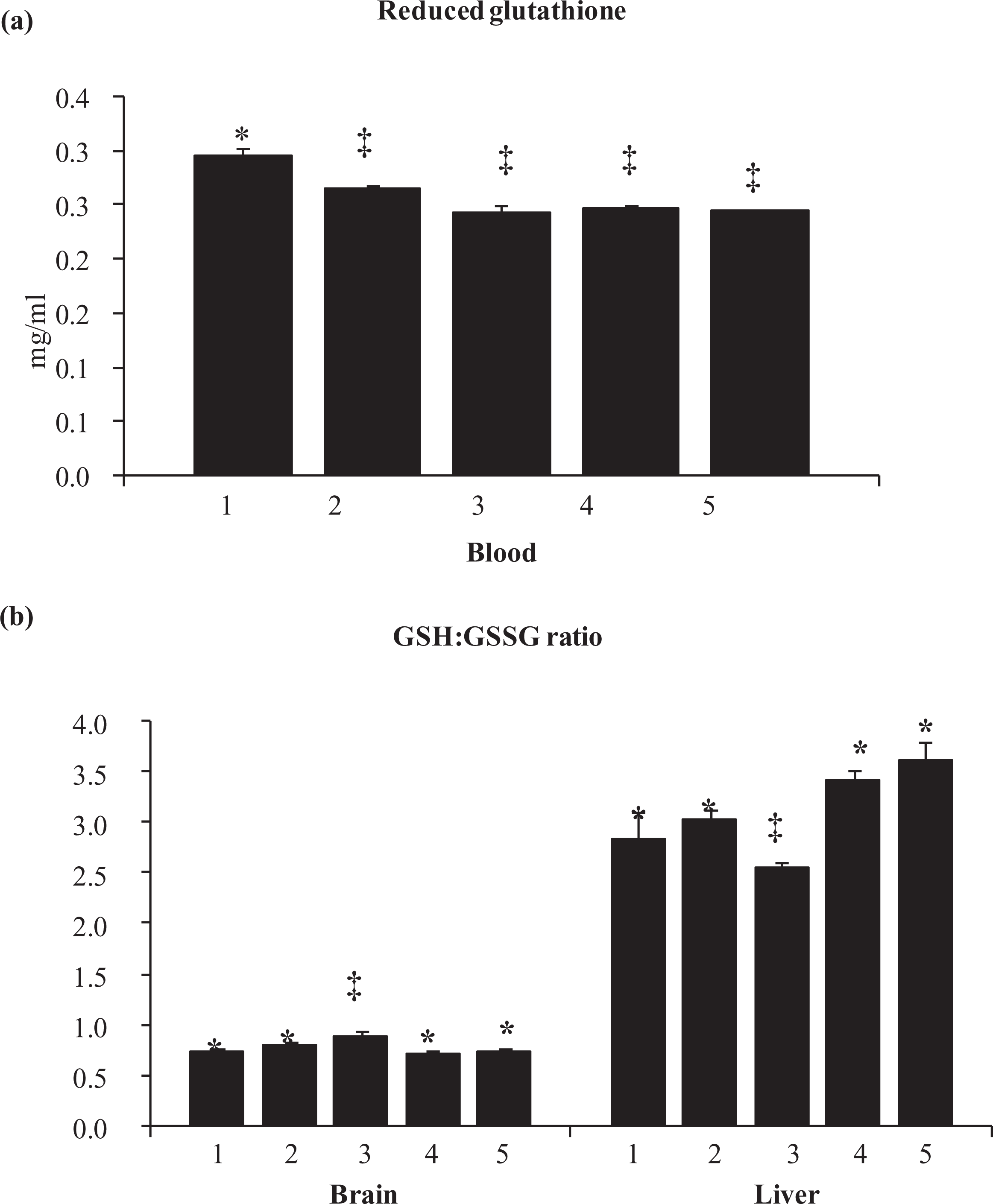
Effect of nicotine pre-exposure on blood reduced glutathione and brain and liver GSH:GSSG ratio of arsenic-exposed Wistar rats. Group: I: control; group 2: nicotine; group III:– arsenic; group IV: nicotine; (0.75 mg/kg) + arsenic, group V: nicotine (3 mg/kg) + arsenic. GSH expressed as milligram per millilitre. Values are mean ± SEM; n = 5. *, ‡: differences between values with matching symbol notations within each column are not statistically significant at 5% level of probability. GSH: reduced glutathione; GSSG: oxidized glutathione.
Effect on GSH:GSSG ratio in tissue
Exposure to nicotine alone at a high dose of 6 mg/kg did not cause any change in the GSH:GSSG ratio in the brain or in the liver (Figure 3). However, arsenic significantly increased and decreased the GSH:GSSG ratio in brain and liver, respectively, compared with control animals. On the other hand, nicotine pretreatment attenuated these effects on GSH:GSSG ratio, in both brain and liver, with an exception of 3 mg/kg nicotine pretreatment that led to a significantly elevated ratio in the liver.
Effect on tissue GST activity
Individually, nicotine and arsenic significantly reduced GST activity in both brain and liver compared with control animals (Figure 4). These effects were more pronounced in the brain exposed to arsenic, while in liver, nicotine caused maximum decrease. However, nicotine pretreatment (both the doses) caused a significant increase in GST activity in both liver and brain compared with arsenic or nicotine alone.

Effect of nicotine pre-exposure on glutathione-S-transferase activity in brain and liver of arsenic exposed Wistar rats. Group I: control; group II: nicotine; group III: arsenic; group IV: nicotine (0.75 mg/kg) + arsenic; group V: nicotine (3 mg/kg) + arsenic. GST expressed as nanomoles of GSH–CDNB conjugate formed per minute per gram of tissue. Values are mean ± SEM; n = 5. *, ‡, §: differences between values with matching symbol notations within each column are not statistically significant at 5% level of probability. GST: glutathione-S-transferase.
Effect on AChE and MAO activity
Figure 5 shows that neither nicotine nor arsenic alone resulted in any alteration in the activity of brain AChE when compared with the control. However, the combination group with high-dose (3 mg/kg) nicotine pretreatment followed by arsenic significantly elevated the brain AChE activity. Figure 5 also indicates that there is no change in brain MAO activity due to arsenic exposure compared to control. However, nicotine alone and the combination groups (nicotine pretreatment followed by arsenic) showed significant reduction in the MAO activity compared with the normal and arsenic-exposed rats.
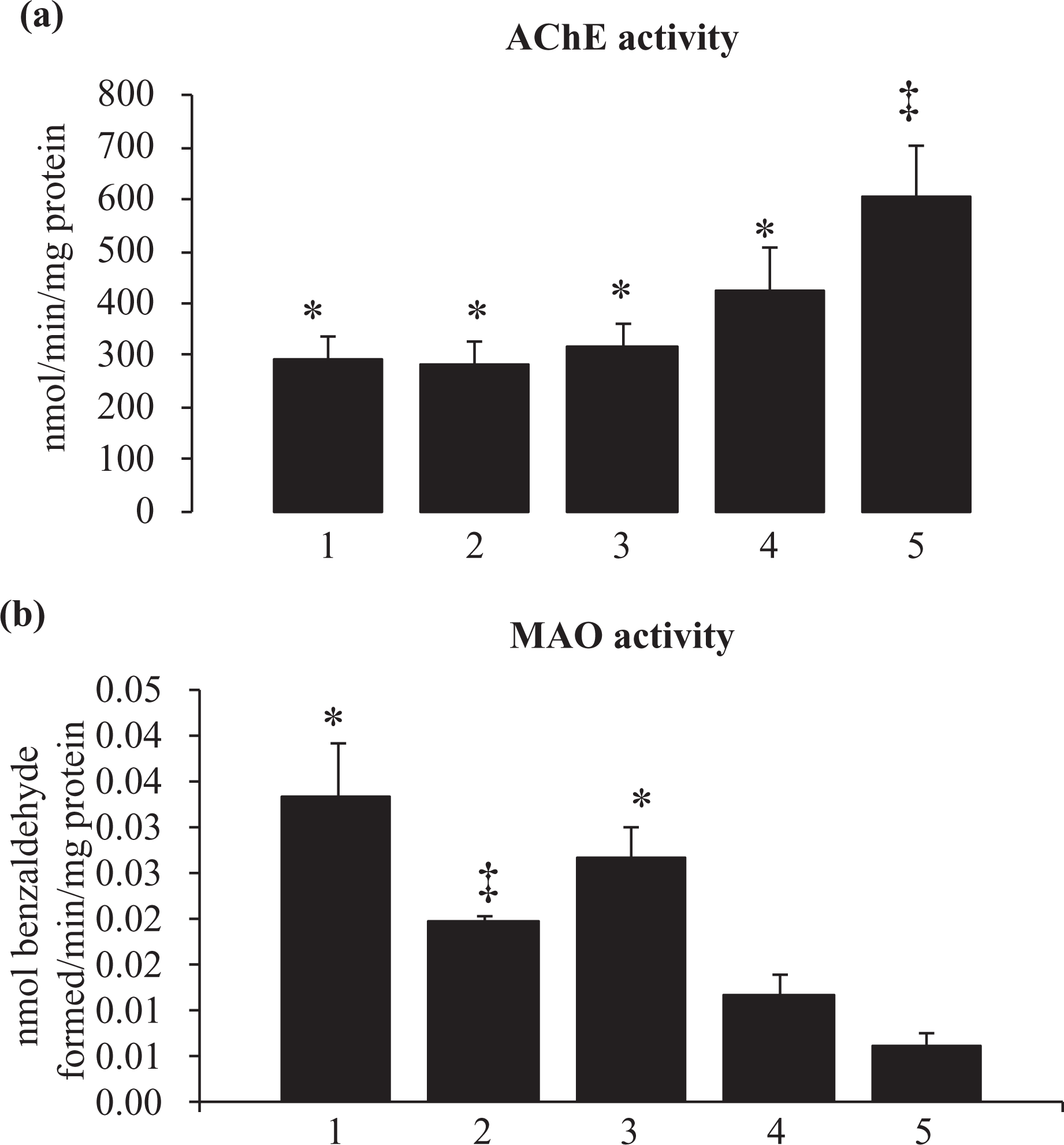
Effect of nicotine pre-exposure on acetylcholinesterase and monoamine oxidase activity in brain of arsenic exposed Wistar rats. Groups I: control; group II: nicotine; group III: arsenic; group IV: nicotine (0.75 mg/kg) + arsenic; group V: nicotine (3 mg/kg) + arsenic. AChE expressed as nanomoles of product formed per minute per milligram of protein; MAO: monoamine oxidase expressed as nanomoles of benzaldehyde formed per minute per milligram of protein. Values are mean ± SEM. *, ‡, §: differences between values with matching symbol notations within each column are not statistically significant at 5%. AChE: acetylcholinesterase.
Discussion
Arsenic is known to cause oxidative stress via generation of free radicals, like ROS and Reactive Nitrogen Species (RNS), and also its metabolic intermediates like dimethyl arsenic (DMA) peroxy radical and DMA radical. 38 –42 On the other hand, nicotine was reported to act as an antioxidant at low doses, while at high doses, it might cause oxidative stress. 43 Nicotine rapidly absorbs and penetrates through physiological barriers including blood–brain barrier. It is extensively metabolized in the liver making it one of the most susceptible organ for its toxic effects. 12
Decreased liver arsenic level in arsenic + nicotine co-exposed animals suggests that nicotine alters arsenic metabolism due to its effect on liver cytochrome. 12 Tobacco smoking and chewing increase the risk of arsenic-induced skin lesions by interfering with arsenic methylation and metabolism. 22 Smoking has been associated with less pronounced arsenic methylation by decreasing conversion of monomethyl arsenic to DMA; 44,45 however, the mechanism is not clear. Smoking inhibits specific As3MT involved in arsenic methylation or impairs one carbon metabolism. 22 Smoking increases serum homocysteine concentration, 46 which via concurrent accumulation of S-adenosylhomocysteine inhibits S-adenosylmethionine-dependent transmethylation reactions. Smoking also lowers folate, vitamin B6 and vitamin B12 essential for homocysteine metabolism. 22,47 Thus, reduced oxidative stress following nicotine pretreatment can be explained by the fact that nicotine is known to alter liver arsenic metabolism. Nicotine pretreatment however increased brain arsenic level, which might be due to altered blood–brain barrier permeability, facilitating arsenic entry. 48
Decreased hepatic ROS due to nicotine (3 mg/kg) pretreatment might be attributed to lower arsenic level (Figure 1), while high (6 mg/kg) nicotine pre-exposure caused an increase in liver ROS supporting the hypothesis that at higher dose it produces oxidative stress. Nicotine and arsenic increased the brain ROS suggesting neurotoxicity. 40,49,50 Increased brain ROS levels during combination of nicotine (3 mg/kg) and arsenic may possibly be due to increased brain arsenic level 12,51,52
Nicotine pretreatment showed protection against arsenic-induced increase in the brain GSH:GSSG ratio. 12,23 However, since nicotine pretreatment caused increase in brain arsenic uptake, protection against oxidative stress cannot be explained. Arsenic-induced decrease in the liver GSH:GSSG ratio may be explained in terms of oxidative stress-mediated depletion of GSH, while protection by nicotine pretreatment might be related to reduced liver arsenic accumulation. High dose of nicotine did not deplete liver GSH levels, which is contrary to earlier reports. 12
GST functions as detoxification enzyme, that is reducing arsenate by mammalian GST Omega. 53 Arsenic- and nicotine-induced inhibition of GST activity in brain and liver might be either due to oxidative stress or direct interaction (GSH conjugation). 54 Arsenic-induced GST inhibition was attenuated by nicotine pretreatment possibly due to modulating arsenic metabolism. Thus, ultimately nicotine-induced alteration in arsenic toxicokinetics may be responsible for its protection against arsenic toxicity.
Arsenic and nicotine also increased liver TBARS levels, while nicotine (3 mg/kg) pretreatment decreased arsenic-induced increase in TBARS suggesting protection by nicotine against arsenic-induced oxidative stress. Brain AChE remained unchanged following nicotine treatment alone, while a significant increase was observed following co-exposure with arsenic. The difference is not completely explained, however, direct acetylcholine (ACh)–receptor binding property of nicotine cannot be overlooked. Nicotine acts as an agonist to brain ACh receptor and may thus increase the release of ACh and other neurotransmitters. 55,56 Increased ACh release may thus increase AChE activity to attain homeostasis. Increased brain arsenic mediated via nicotine pretreatment may be an additional factor for this effect. Inhibition of MAO in nicotine alone and pre-exposed arsenic-treated rats might be due to direct scavenging of radicals produced due to MAO-B activity or by directly inhibiting MAO after binding. 45,57,58
Contrary to the earlier reports on synergistic toxic interaction between arsenic and tobacco, we report some prophylactic efficacy of nicotine against arsenic toxicity. Thus, the possible toxic synergism reported earlier may be between arsenic and tobacco constituent/constituents other than nicotine. Nicotine-induced alterations in the brain and liver arsenic level are interesting findings that require further exploration. Furthermore, measuring brain and tissue nicotine levels may also reveal important toxicokinetics pattern, which was a limitation in the present study.
Footnotes
Authors’ Note
Authors declare that this manuscript was consistent with the guidelines and principles of the American College of Toxicology Statement on the Use of Animals of Toxicology.
Acknowledgements
Authors thank Director of the establishment for his support and encouragement.
Conflict of interest
The authors declared no conflicts of interest.
Funding
The author V.P. was awarded Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), New Delhi, India.