
Abstract
Objective:
The aim of the present study was to observe the effects of 1.8 GHz radiofrequency (RF) radiation on the protein expression of human lens epithelial cells (hLECs) in vitro.
Methods:
The hLECs were exposed and sham-exposed to 1.8 GHz RF radiation (specific absorption rate (SAR) of 4 W/kg) for 2 h. After exposure, the proteins extracted from LECs were loaded on the Ettan MDLC system connected to the LTQ-Orbitrap MS for screening the candidate protein biomarkers induced by RF. The quantitative real-time polymerase chain reaction (qRT-PCR) was used to detect the levels of messenger RNA of candidate biomarkers. After the hLECs were exposed to 1.8 GHz RF (SAR of 2, 3 and 4 W/kg) for 2 h, the Western blot assay was utilized to measure the expression levels of the above-screened candidate protein biomarkers.
Results:
The results of shotgun proteomic analysis indicated that there were eight proteins with differential expression between exposure and sham exposure groups. The results of qRT-PCR showed that there were three genes with expressional differences (valosin containing protein (VCP), ubiquitin specific peptidase 35 (USP35) and signal recognition particle 68 kDa (SRP68)) between exposure and sham exposure groups. The results of Western blot assay exhibited that the expressional levels of VCP and USP35 proteins significantly increased and the expressional level of protein SRP68 significantly decreased in hLECs exposed to 1.8 GHz RF radiation (SAR of 3 and 4 W/kg) for 2 h when compared with the corresponding sham groups (p < 0.05).
Conclusion:
The shotgun proteomics technique can be applied to screen the proteins with differential expression between hLECs exposed to 1.8 GHz RF and hLECs sham-exposed to 1.8 GHz RF, and three protein biomarkers associated with RF radiation were validated by Western blot assay.
Introduction
The widespread use of mobile telecommunications has aroused public concern with potential health risks associated with exposure to radiofrequency (RF) electromagnetic fields from these novel products of mobile phones. A variety of studies have already been performed on the possible biological and physical effects of mobile phone radiation. Some in vitro studies demonstrated the effects of RF on DNA damage, chromosome aberration, cell cycle distribution, cell proliferation, cell survival, stress response and so on, but still a lot of biological effects induced by RF are unknown, for example, the protein expression changes induced by RF, 1 and the high-throughput screening proteomics techniques can be used to identify a variety of potential molecular targets associated with RF radiation. 2 The potential molecular targets obtained by proteomic analysis might be used in further investigations to ascertain the possible alteration of expression levels of protein, which are related to the biological effects induced by RF radiation and to understand the possible biochemical mechanism of harmful effects.
In our previous studies, it was successfully found that the changes in the expression level of the heat shock protein 70 (HSP70) and heterogeneous nuclear ribonucleoprotein K (hnRNP K) proteins of human lens epithelial cells (hLECs) exposed to RF were detected using a popular proteomic technology, that is, two-dimensional electrophoresis (2-DE) combined with mass spectrometry, 3 and HSP70 and hnRNP K proteins were involved in the stress reaction. It has also been reported that the mobile phone radiation with non-thermal effect could induce changes in the expression level of HSP27 protein and p38 mitogen-activated protein kinase in EA.hy926 human endothelial cells, according to the results of 2-DE. 4 The same study group provided the lists of genes and proteins with significant expression changes in the EA.hy926 and the EA.hy926v1 variant, 5 and identified eight proteins that could be significantly affected on forearm’s skin in human volunteers exposed to RF radiation. 6 Other study groups reported the negative effects of RF radiation on protein expression profiles in MCF7 cells. 1 ,7 However, the data about proteomics studies associated with RF are still limited, moreover, until now, the proteomics experiments associated with RF have been based on 2-DE for separating proteins. There are some disadvantages of the 2-DE technique, which include a large amount of sample handling, a limited dynamic range and the difficulties in resolving low-abundance proteins with extreme isoelectric point and molecular weight. 8 These issues led to the development of alternative solutions for proteome analysis, that is, the development of shotgun proteomics technique. In a generalized shotgun proteomic pipeline, a mixture of proteins is digested into peptides that are loaded onto at least a two-dimensional chromatography-based separation system, the peptides are eluted into a tandem mass spectrometer (MS/MS) in an automated fashion and the resulting tandem mass spectrometry data are analyzed by powerful computational systems. 8 Shotgun proteomic platform is a robust alternative of gel-based approaches for analyzing the complex protein samples.
Considering the limited data of changed expressional levels of lens proteins induced by RF and the development of new proteomic analysis techniques, in the present in vitro investigation, the high-throughput technique of shotgun proteomics was applied to screen the proteins with the expressional changes in hLE-B3 cells exposed to 1.8 GHz RF (specific absorption rate (SAR) of 4 W/kg) for 2 h, then the differentially expressed proteins induced by RF were validated by Western blot assay. The aim of the present investigation was to find the new differentially expressed proteins of hLECs exposed to RF, which may be related to pathological changes in human lens exposed to RF.
Materials and methods
Cell culture
hLE-B3 cells were obtained from Marjorie F. Lou (University of Nebraska Medical Center). The cells were cultured in Dulbecco’s modified Eagle medium (Gibco, New York, New York, USA) containing 20% fetal bovine serum (Gibco, New York, New York, USA) at 37°C in a humidified atmosphere of 5% CO2. The cells were cultured in a 35 mm dish (NUNC, Roskilde, Denmark) containing a total volume of 2 ml medium. The cells were divided into three exposure groups that are continuingly exposed to 1.8 GHz RF at a SAR of 2, 3, or 4 W/kg, respectively, for 2 h and the corresponding sham exposure groups.
Exposure system
The RF exposure system (sXc-1800 system) with a GSM signal was designed by the Foundation for Information Technologies in Society (IT’IS, Zurich, Switzerland). It mainly consists of an RF generator, an arbitrary function generator, a narrow band amplifier and two rectangular waveguides operating at a frequency of 1.8 GHz. The two waveguides, one for exposure and the other for sham exposure, are placed inside a conventional incubator to ensure constant environmental conditions (37°C, 5% CO2/95% air atmosphere). The increased temperatures of the cells within the culture dishes exposed to the SAR of 2, 3 and 4 W/kg were 0.054, 0.081 and 0.108°C, respectively. A dish holder inside the waveguide guarantees that the dishes are placed exactly in the H-field maximum of the standing wave and exposed simultaneously in E polarization inside a waveguide. The system enables the exposure of a monolayer of cells with less than a 30% non-uniformity of SAR. Six Petri dishes can be exposed simultaneously in one exposure waveguide. The entire setup is controlled by a computer, enabling automated control of the exposure parameters including exposure strength (SAR), exposure time and exposure pattern. The RF electromagnetic field (EMF) simulating the GSM 1.8 GHz signal is amplitude-modulated by a rectangular pulse with a repetition frequency of 217 Hz and a duty cycle of 1:8. The RF exposure pattern was continuous.
Liquid chromatography MS/MS
Sample preparation and analysis of nano-LC-MS/MS
After exposure and sham exposure to a 1.8-GHz RF (SAR of 4 W/kg) for 2 h, the cells were washed twice with cold 0.01 M phosphate-buffered saline (PBS), scraped into 1 ml of 0.01 M PBS, pelleted and lysed in the Mammalian Protein Extraction Reagent (Thermo, Rockford, Illinois, USA) for 30 min at 4°C, then subjected to centrifugation at 14,000 r/min for 30 min at 4°C. The concentration of total cell proteins was assayed by a standard Bradford protein assay (Bio-Rad, Richmond, California, USA). 9
In-solution trypsin digestion
The samples were precipitated by acetone and dissolved in reducing solution (8 M urea; Sigma, St Louis, Missouri, USA), and 100 µg of protein sample for each fraction was reduced with 10 mM dithiothreitol (DTT) (Sigma) at 37°C for 45 min and alkylated with 50 mM iodoacetamide (Sigma) at room temperature for 45 min in the darkness. Then the alkylation was stopped by 40 nM DTT solution and the protein sample was incubated in the solution at room temperature for 45 min. After dilution in a solution of 50 mM ammonium bicarbonate (NH4HCO3) (Sigma), the protein mixture was digested by sequencing grade-modified trypsin (Promega, Madison, Wisconsin, USA) using a 1:50 enzyme/protein ratio at 37°C for 20 h. The tryptic peptide mixture was lyophilized and kept at −80°C until use.
Mass spectrometric analysis
Tryptic peptides were resuspended in 0.1% formic acid and loaded onto an Ettan MDLC system (GE Healthcare, Pittsburgh, Pennsylvania, USA) connected to an LTQ-Orbitrap MS (Thermo Finnigan, Bremen, Germany). The separation of the resulting tryptic peptide mixtures was performed by nanoscale reverse-phase liquid chromatography (LC). The Ettan MDLC nanoflow/capillary LC system (GE Healthcare) was equipped with a trap column (Dionex/LC Packings μ-Precolumn Cartridge P/N 160454 C18 PepMap 100, 5 μm, 100 Å, 300 mm internal diameter (i.d.) × 5 mm) and a nanocolumn (Dionex/LC Packings P/N 160321, 150 × 0.075 mm i.d., C18 PepMap, 3 μm, 100 Å). The separated peptides were transferred into an LTQ-Orbitrap (Thermo Finnigan) with a nano ion spray source. The full scan MS spectra (m/z 300–4000) were acquired in the orbitrap with the resolution at R = 60,000 at m/z 400 with the number of accumulated ions being 1 × 106. The five most intense ions were isolated and fragmented in a linear ion trap. The resulting fragment ions were recorded by the LTQ.
MS data analysis
The MS data were analyzed using X!Tandem. The raw data acquired by the MS above were submitted to X!Tandem along with a FASTA formatted file of the protein database ENSEMBL containing protein data of human.
In the X!Tandem search parameter setting, the carboxyamidomethyl cysteine (Cys-CAM) was included as a fixed modification of 57.0215 Da for iodoacetamide reduction and alkylation. The cysteines might react with free acrylamide monomers to form propionamide cysteines (Cys-PAM). An optional 14 Da was included in the search to account for potential Cys-PAM, as the mass difference between Cys-PAM and Cys-CAM is 14 Da. Methionine residue modification was accounted for by the inclusion of an optional 15.9949 Da. The peptides were searched using partial tryptic cleavage constraints and up to one missed-cleavage sites were allowed for tryptic digestion. For the X!Tandem search, the mass tolerances were 0.5 Da for parent masses and 100 ppm for fragment masses.
The trans-proteomic pipeline (TPP) (http://tools.proteomecenter.org) is a collection of integrated tools for MS/MS proteomics. The v4.0 JETSTREAM rev 2 version of TPP was used in this study. TPP includes PeptideProphet and ProteinProphet, two statistical programs for proteomics data analysis. The RAW format data obtained by the LTQ-Orbitrap were converted to the mzXML format by the TPP Web-interface tool. The putative proteins were annotated by Global Proteome Machine (GPM) and the controlled false discovery rate (FDR) of GPM was 5%.
Chemometrics analysis
The spectral counting was performed as described previously. 10 The MS/MS spectral data were analyzed with the SIMCA-P 12.0 (Umetrics AB, Sweden) for the multivariate data analysis. Before multivariate data statistics analysis, the data were mean-centered and pareto-scaled. The unsupervised principal component analysis (PCA) was first utilized in all the samples to visualize the general clustering and find the outlying data. Then the supervised partial least squares latent structure discriminate analysis (PLS-DA) was performed to identify the biomarkers that contributed to the clustering observed in the PCA. The R 2 Y as a parameter indicates the total explained variation in the X matrix. Q2 represents the high predictability of the model. Potential biomarkers of differentiating sham-groups from RF groups were selected according to the variable importance in the projection (VIP) values. The VIP values reflect the influence of each protein on the different groups.
Quantitative real-time polymerase chain reaction
After exposure and sham exposure to 1.8 GHz RF (SAR of 4 W/kg) for 2 h, the cells were washed twice with cold PBS and the total cellular RNA was isolated from hLECs using the TRIzol reagent (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s protocol. The concentration of RNA was determined spectrophotometrically at 260 nm. The quality of the RNA was determined on 1% agarose gels. RNA was reverse transcribed using the PrimeScript RT-PCR System kit (TaKaRa, Tokyo, Japan) in the presence of oligo-dT primers, 0.045 μg total RNA was reverse transcribed with 5× PrimeScript® buffer, 50 μM oligo-dT primer, 50 μM random 6 mers and PrimeScript reverse transcriptase mix I. The reaction mixture was incubated at 37°C for 15 min, 85°C for 5s and held at 4°C. The products were subjected to real-time polymerase chain reaction (RT-PCR) analysis using a SYBR® Premix Ex Taq™ Mix (TaKaRa) with an ABI PRISM 7500 sequence detection system (Applied Biosystems, Grad Island, New York, USA). The specific PCR primers for differentially expressed candidate genes and the internal control gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were designed with the Primer Premier 3 (Primer, Canada) and Oligo 6.0 (Molecular Biology Insights, Cascade, Colorado, USA; Table 1). Template-negative control experiments were carried out to exclude any possibility of DNA contamination in primers. The 20 μl PCR reactions were conducted using 1 μl of complementary DNA (cDNA), 10 μM of the forward and reverse primers, 10 μl 2× SYBR Premix Ex Taq and 0.4 μl 50× ROX reference dye. The cycling parameters were an initial cDNA denaturation step at 95°C for 30s followed by 40 cycles of PCR with cDNA denatured at 95°C for 3s and extension at 60°C for 30s. The fluorescence was measured at 60°C. The dissociation curves of PCR products were obtained to guarantee amplification of the correct genes. The Ct values (threshold cycle, marking the cycle when the fluorescence of a given sample significantly exceeded the baseline signal) were computed by the SDS software (version 2.0; Applied Biosystems). ▵Ct sample values were calculated as Cttarget gene − CtGAPDH. The N-fold increase or decrease in expression was calculated by the ▵▵Ct method with the sham group Ct values as the reference point. N-Fold difference was determined as 2−(▵Ct RF group − ▵Ct sham group). In two groups, six genes including valosin containing protein (VCP), signal recognition particle 68 kDa (SRP68), ubiquitin specific peptidase 35 (USP35), cytochrome P450 21 (CYP21A2), tubulin alpha 1b (TUBA1B) and the internal control GAPDH were detected in each sample. Three replicate reactions were performed for each sample and the average ▵Ct was calculated.
The oligonucleotides served as the primers of qRT-PCR.

CYP21A2: cytochrome P450 21; SRP68: signal recognition particle 68 kDa; VCP: valosin containing protein; USP35: ubiquitin specific peptidase 35; TUBA1B: tubulin alpha 1b; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; qRT-PCR: quantitative reverse transcriptase polymerase chain reaction.
Western blot assay
After exposure and sham exposure to 1.8 GHz RF (SAR of 2, 3 and 4 W/kg) for 2 h, the cells were treated according to the above sample preparation of MS. Equal amounts of protein samples (20 µg) were separated on a 10% SDS-PAGE gel, transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, Massachusetts, USA), then probed with various primary antibodies including rabbit polyclonal anti-SRP68 antibody (catalog number 11581-1-AP, Proteintech, Chicago, Illinois, USA), rabbit polyclonal anti-GAPDH antibody (catalog number sc-25778, Santa Cruz Biotechnology, Santa Cruz, California, USA), rabbit polyclonal anti-USP35 antibody (catalog number ab86791, Abcam, Cambridge, Massachusetts, USA) and rabbit monoclonal anti-VCP antibody (catalog number 3339-1, Epitomics, Burlingame, California, USA) overnight at 4°C followed by fluorescence-labeled secondary antibody (Li-COR, Lincoln, Nebraska, USA) diluted 1:5000 in Tris Buffered Saline with Tween-20 (TBST) for 1 h at room temperature. Finally, blots were developed with the Odyssey system (Li-COR). The protein expression was analyzed using the ImageJ program (Wayne Rasband, NIH, Maryland, USA). The target protein expression was normalized with respect to GAPDH expression.
Statistical analysis
Each experiment was repeated at least three times. Statistical analyses were conducted with SPSS software version 16.0 (SPSS Inc., Chicago, Illinois, USA).The differences in data between RF exposure groups and corresponding sham exposure groups were analyzed with the independent Student’s t test. The statistical threshold was set at 0.05 (two-sided).
Results
Differential protein expression analysis by label-free shotgun proteomics
LC-MS/MS analysis of the protein samples for sham exposure group and RF exposure group (SAR of 4 W/kg) revealed a total of 462 nonredundant proteins based on the identification of two or more unique peptides. Of these, 344 proteins were identified in both groups, and a total of 406 and 400 proteins were identified in sham exposure and RF exposure groups, respectively. Using a randomized database as a decoy database, the FDR for the protein identification at 0.67% was estimated. The low FDR highlights the fidelity of the current search approach.
Label-free quantitative comparison between the two groups depends on the PLS-DA module. The MS/MS spectra of two groups were successfully analyzed using PLS-DA. The model had the high ability of explanation and prediction (R 2 Y = 0.999, Q2 = 0.925). Table 2 lists the results obtained by LC-MS/MS analysis, which indicate that the VIP values of 14 proteins are higher than 2, and there were significant differences in expression levels in eight proteins between the sham exposure group and RF exposure group (p < 0.05). Among the eight proteins, the TUBA1B and VCP had high VIP values, the CYP21A2 and USP35 had the highest fold changes in differential protein expression levels between sham exposure group and RF exposure group, the SRP68 was the only downregulated protein after RF radiation. So, the above five proteins were considered as candidate proteins for the next step of the experiment.
The list of proteins with VIP values higher than 2.

VIP: variable importance in the projection; RF: radiofrequency.
a p < 0.05, When compared with the corresponding sham exposure group.
The screening results at the transcriptional level
The expressional levels of RNA of VCP, SRP68, USP35, CYP21A2 and TUBA1B in hLECs exposed to1.8 GHz RF (SAR of 4 W/kg) for 2 h are shown in Figure 1. GAPDH was used as an internal control. It was found that the significant upregulation of VCP and USP35 was detected (p < 0.01) and the significant downregulation of SRP68 was found (p < 0.05) in the RF exposure groups, when compared with sham exposure groups. But there were no significant differences in expression levels of RNA of CYP21A2 or TUBA1B between RF exposure groups and corresponding sham exposure groups (p > 0.05).
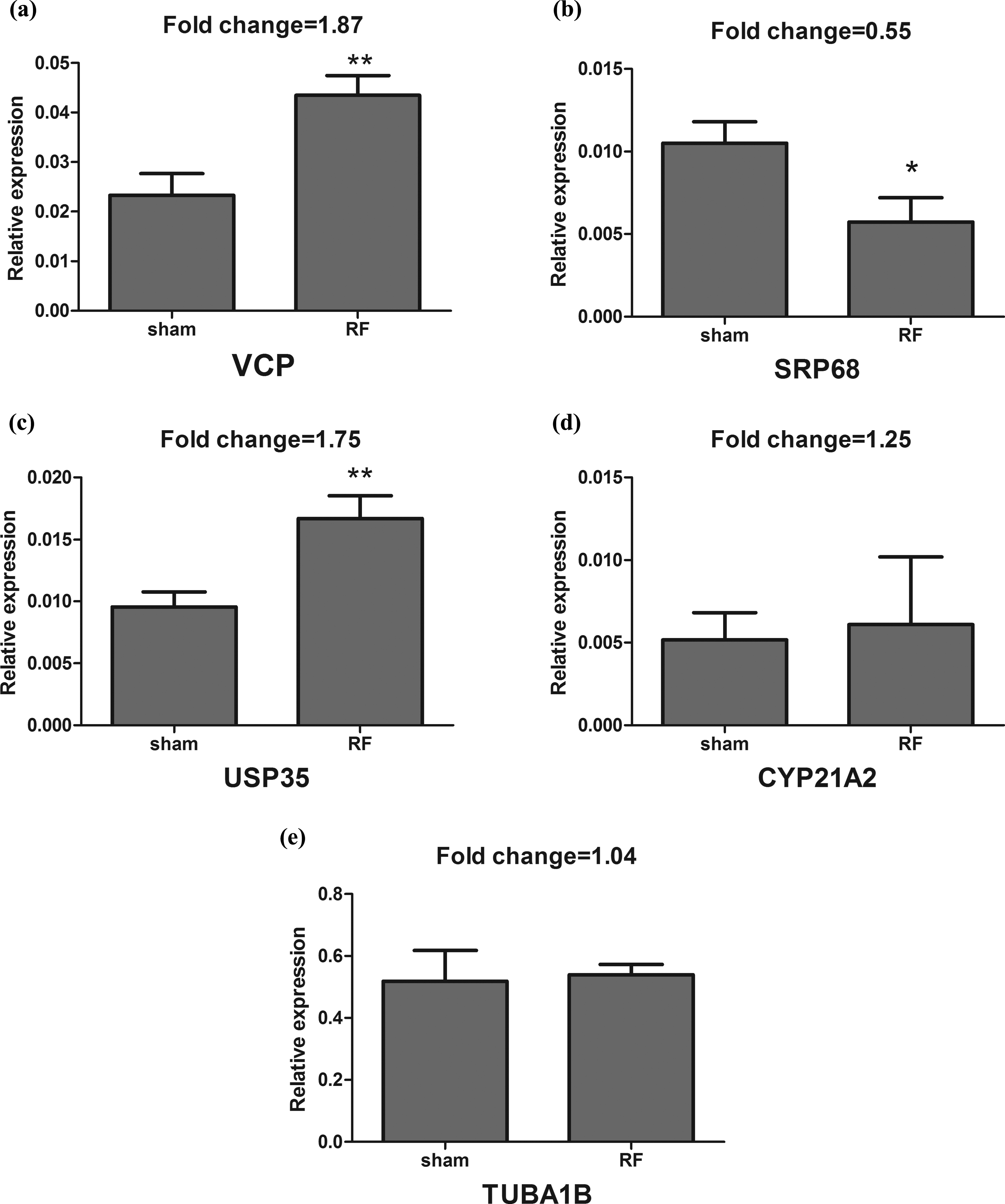
The expression levels of mRNA for VCP (a), SRP68 (b), USP35 (c), CYP21A2 (d) and TUBA1B (e) were examined by qRT-PCR. The expression of GAPDH mRNA was used as the internal standard. *p < 0.05, when compared with the corresponding sham exposure groups; **p < 0.01, when compared with the corresponding sham exposure groups. mRNA: messenger RNA; VCP: valosin containing protein; SRP68: signal recognition particle 68 kDa; USP35: ubiquitin specific peptidase 35; CYP21A2: cytochrome P450 21; TUBA1B: tubulin alpha 1b; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; qRT-PCR: quantitative reverse transcriptase polymerase chain reaction.
The expressional levels of proteins validated by Western blot assay
The expressional levels of three proteins (VCP, SRP68 and USP35) in hLECs exposed to 1.8 GHz RF (SAR of 2, 3 and 4 W/kg) for 2 h are shown in Figures 2 to 4. GAPDH served as a loading control. Figure 2 indicates that the expression levels of VCP in hLECs exposed to 1.8 GHz RF (SAR of 3 and 4 W/kg) for 2 h significantly increased when compared with the corresponding sham exposure groups (p < 0.05). Figure 3 exhibits that the expression levels of SRP68 in hLECs exposed to 1.8 GHz RF (SAR of 3 and 4 W/kg) for 2 h significantly decreased when compared with the corresponding sham exposure groups (p < 0.05). Figure 4 shows that the expression levels of USP35 in hLECs exposed to 1.8 GHz RF (SAR of 3 and 4 W/kg) for 2 h significantly enhanced when compared with the corresponding sham exposure groups (p < 0.05).

(a) Western blot assay of VCP and loading control (GAPDH). (b) The expression levels of VCP in hLECs exposed to RF (SAR of 2, 3 and 4 W/kg) for 2 h were detected by Western blot assay. *p < 0.05, when compared with the corresponding sham exposure groups. VCP: valosin containing protein; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; hLECs: human lens epithelial cells; RF: radiofrequency; SAR: specific absorption rate.

(a) Western blot assay of SRP68 and loading control (GAPDH). (b) The expression levels of SRP68 in hLECs exposed to RF (SAR of 2, 3 and 4 W/kg) for 2 h were detected by Western blot assay. *p < 0.05, when compared with the corresponding sham exposure groups. SRP68: signal recognition particle 68 kDa; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; hLECs: human lens epithelial cells; RF: radiofrequency; SAR: specific absorption rate.

(a) Western blot assay of USP35 and loading control (GAPDH). (b) The expression levels of USP35 in hLECs exposed to RF (SAR of 2, 3 and 4 W/kg) for 2 h were detected by Western blot assay. *p < 0.05 when compared with the corresponding sham exposure groups. USP35: ubiquitin specific peptidase 35; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; hLECs: human lens epithelial cells; RF: radiofrequency; SAR: specific absorption rate.
Discussion
Although some studies about the health effects induced by RF radiation have been reported, there are much more diverse issues of RF radiation that remain unclear. In 2001, Leszczynski et al. proposed and subsequently demonstrated that proteomics could be used as a tool for detecting the changes in protein expression caused by RF radiation. 11,12 However, so far the proteomics approach has been used only in a few studies for examining the effects of the RF radiation. 1,5,6,13 On the basis of the development of the proteomics technique, a new hypothesis that the possible biological effects induced by RF radiation could be tested using new proteomics techniques has been suggested. Li et al. used 2-DE combined with mass spectrometry to measure the hLECs exposed to RF and found that HSP70 and hnRNP K were involved in the stress reaction induced by RF. 3 Therefore, according to the limited data it is difficult to obtain the general conclusion about the effects induced by RF on cellular proteome and the physiological processes regulated by those influenced proteins, including human lens pathogenesis.
In this study, the total protein expression of hLECs exposed to RF- or sham-radiation in vitro was studied by the LTQ-Orbitrap system, which is a new high-performance liquid chromatography-MS/MS system, and the system for high mass accuracy has been widely adopted. The coupling of this system to shotgun proteomic approaches promises to provide the larger scale datasets than those achieved previously. 8 Although, the 2-DE technique as a classical proteomics approach can resolve thousand spots on a two-dimensional protein map, the number of intracellular proteins may be more than 30,000. In fact, the 2-DE technique only is able to detect protein spots with abundant expression. On the other hand, the false-positive results may appear due to the problems in reproducibility and variability between experiments. The results of the present investigation indicated that the shotgun proteomics approach can overcome these shortages and provide a convenient and reliable method for comparative proteomics studies. In the present experiment, the changed expression levels of three proteins (VCP, SRP68 and USP35) in hLECs exposed to RF radiation were detected with this approach. These results were also successfully validated by quantitative RT-PCR (qRT-PCR) at the transcriptional level and Western blot assay at the protein level.
Other authors reported that VCP (also known as Cdc48 or p97) is a conserved hexameric AAA+ (ATPases associated with diverse cellular activities) type ATPase. 14 VCP comprises a substrate and cofactor binding N domain followed by two AAA ATPase domains termed D1 and D2 and forms a hexameric double-ring structure. VCP plays critical roles in a broad range of diverse cellular processes, including Golgi, endoplasmic reticulum (ER) and nuclear membrane reassembly of ER-associated degradation (ERAD), the ubiquitin (Ub)–proteasome system (UPS), cell cycle regulation, DNA repair, preventing polyglutamine aggregation, autophagosome maturation and mitophagy. 14 Functional diversity of VCP is mainly determined by differential binding of distinct cofactors/adaptor proteins. In a recent study, it was found that genetic inactivation of VCP increased the levels of misfolded rhodopsin in Drosophila retinas, 15 which suggested that VCP is a Ub-selective chaperone and that its key function is to unfold proteins and disassemble protein complexes. The cellular stress response is a reaction of cells to environmental stimuli, such as heavy metals, heat shock, anoxia and RF and so on. In response to weak environmental stresses, the cells are able to maintain homeostasis by the stress response pathway, such as the activation of HSP. 16,17 VCP, as a structural protein, forms a complex with clathrin and HSP70. 18 The chaperone ability of VCP is to prevent unfolded proteins from aggregating and precipitating out of solution. 19 VCP cooperates with the Ub ligase RNF8 to orchestrate the assembly of signaling complexes and the efficient DSB repair after exposure to ionizing radiation. These findings consider VCP as an essential factor in Ub-governed DNA damage response and highlight the importance of VCP in guarding genome stability. 20 In the present work, the upregulation of VCP suggests that RF exposure can induce the stress response in hLECs.
Other authors also reported that the UPS plays central roles in protein quality control. Ub is a highly conserved protein and is involved in the post-translational protein modification in eukaryotes. Ubiquitination occurs on lysine residues of substrates through the cooperative activity of Ub activating (E1), Ub conjugating (E2) and Ub ligase (E3) enzymes. Short-lived proteins and misfolded and damaged proteins should be appropriately degraded by the UPS pathway. Ubiquitination is opposed by the action of deubiquitinating enzymes (DUBs). E3s are counteracted by DUBs, which are proteases that serve to deconjugate the Ub-modified substrates. In case of DUBs, their function is to rescue substrate proteins from proteasomal degradation, to recycle Ub or even to control protein trafficking. 21 USP35 is a kind of DUBs but VCP is an Ub-selective chaperone. It has been demonstrated that there is an interaction between VCP and E3s, gp78 or Hrd1, which plays an important role in ERAD. ERAD also plays a role in protein quality control. In the present work, the alteration of VCP in concert with USP35 suggested that the quality control systems could keep the homeostasis of hLECs exposed to RF radiation. Therefore, the increased expressional levels of VCP and USP35 proteins in hLECs exposed to RF radiation may be a protective response to RF exposure for 2 h.
The SRP is a ribonucleoprotein complex that transports the secreted membrane proteins to the ER for processing. 22 The complex includes a 7S RNA and six different polypeptides of 9, 14, 19, 54, 68 and 72 kDa. 22 SRP is a key component of the cellular machinery that couples the ongoing synthesis of proteins to their proper localization. 23 Among the six polypetides of the eukaryotic SRP, SRP68 and SRP72, two largest polypetides, form a stable SRP68/72 heterodimer, which is required for SRP function. Sorting of secretory proteins is a fundamental cellular function in which the SRP plays a central role. 24 After exposure to RF, the downregulation of SRP68 protein indicated that RF radiation may affect the protein transportation in hLECs.
Conclusion
The shotgun proteomics technique can be applied to screen the differentially expressed proteins in hLECs exposed to 1.8 GHz RF and three protein biomarkers associated with RF radiation were validated by Western blot assay. The upregulated VCP and USP35 proteins may be involved in the protein quality control reaction of hLECs exposed to RF. The downregulation of SRP68 protein indicated that RF may affect the protein secretion.
Footnotes
Funding
The present study was supported by National Nature Science Foundation of China (30900273), Foundation Project for Medical Science and Technology of Zhejiang Province (2009A101), Zhejiang Key Laboratory Fund of China (2011E10006) and Zhejiang Key Innovation Team Project of China (2009R50039).
Acknowledgments
The authors would like to express their gratitude to Professor Xu Zhengping of Zhejiang Provincial Key Bioelectromagnetics Laboratory in Zhejiang University for providing the 1.8 GHz RF source.