
Abstract
Aluminum phosphide (ALP), a widely used fumigant and rodenticide, leads to high mortality if ingested. Its toxicity is due to phosphine that is liberated when it comes in contact with moisture. The exact site or mechanism of action of phosphine is not known, although it is widely believed that it affects mitochondrial oxidative phosphorylation. Basic serum biochemical parameters, activity of mitochondrial complexes, antioxidant enzymes and parameters of oxidative stress were estimated in the platelets of 21 patients who developed severe poisoning following ALP ingestion. These parameters were compared with 32 healthy controls and with 22 patients with shock due to other causes (cardiogenic shock (11), septic shock (9) and hemorrhagic shock (2)). The serum levels of creatine kinase-muscle brain and lactate dehydrogenase were higher in patients poisoned with ALP, whereas a significant decrease was observed in the activities of mitochondrial complexes I, II and IV. The activity of catalase was lower but the activities of superoxide dismutase and glutathione peroxidase were unaffected in them. A significant increase in lipid peroxidation and protein carbonylation was observed, whereas total blood thiol levels were lower. In patients severely poisoned with ALP, not only cytochrome c oxidase but also other complexes are involved in mitochondrial electron transport, and enzymes are also inhibited.
Keywords
Introduction
Aluminum phosphide (ALP) is widely used as a grain fumigant and rodenticide in India 1 and is available in markets predominantly as granulated powder. The easy availability makes it a commonly used compound for self-harm in India, especially in north, northwestern and central parts of the country. 2 Sporadic cases of ALP exposure have been reported from all over the world and extensive suicidal poisoning has been reported from Iran 3 in addition to India. Following ingestion, on contact with moisture in the gut it gets hydrolyzed, releasing phosphine which is highly toxic. However, subsequent toxicodynamics and the exact mechanism of the action of phosphine are not clear. The management of ALP poisoning continues to be unsatisfactory as there is no specific antidote and mainly supportive measures to treat shock and hypotension are being offered.
Toxicology of phosphine has been explored in a variety of organisms, but human data are comparatively less. The available few reports in insects and rats indicate that it exerts its toxic effects by interfering with mitochondrial energy production and oxidative stress generation. 4 –8 Human data available at present is mostly confined to its clinical presentation, complications and outcome. The pathophysiologic mechanisms are yet to be studied in humans. An earlier study has observed a decrease in the activity of cytochrome c oxidase (COX) in patients with severe ALP poisoning. 1 The present study was undertaken to evaluate the serum biochemical profile, activities of mitochondrial complexes (I, II and IV), activity of antioxidant enzymes (catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPX)) and markers of oxidative stress in patients with significant ALP poisoning.
Methods
Study protocol
The study was performed at a tertiary care center situated in north-west India, about 250 km north of Delhi. The study was approved by the Institute ethics committee. Patients above 18 years of age of both the sexes who were admitted to the emergency department within 6 h of ALP ingestion and who had significant poisoning were included in ALP group. Patients with insignificant poisoning (who did not develop hypotension and shock after ingestion) or had other comorbidities or if there was doubtful history of ingestion were excluded. The patients attending medical outpatient for health checkup and found to be healthy were recruited as positive controls. Patients with shock due to other causes were included as negative controls to rule out the possibility of shock per se causing inhibition, as COX inhibition has also been reported to occur in hemorrhagic and septic shock. 9
Informed consent was obtained from the subjects or their close relatives; 10 ml of venous blood was drawn from the antecubital vein of the patient. From this, 7 ml was transferred into tubes containing acid citrate dextrose buffer (pH 6.8) and the platelets were extracted according to the method of Slichter and Harker. 10 The resultant plasma was used for the assay of protein carbonylation and lipid peroxidation. From 10 ml of blood, 3 ml was used for estimating other serum biochemical parameters and total blood thiol levels.
Chemicals
Cytochrome c, 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB), nicotinamide adenine dinucleotide (NADH), reduced glutathione, bovine serum albumin and nitro blue tetrazolium were purchased from Sigma Chemical Co. (St Louis, Missouri, USA). Magnesium sulfate, sodium carbonate, sucrose, sodium chloride and copper sulfate were purchased from Qualigens (India). Kits for biochemical assays were obtained from Roche Diagnostics K.K. (Tokyo, Japan). The biochemical parameters in serum were assayed using Hitachi Modular P800 autoanalyzer (Roche Diagnostics K.K.) after calibration and quality control. Protein concentration in platelet sample was determined by Lowry’s method using bovine serum albumin as the standard. 11 Activities of mitochondrial complexes I, II and IV, antioxidant enzymes (CAT, SOD and GPX) and markers of oxidative damage were estimated by standard photometric methods using DU 640 UV spectrophotometer (Beckman Coulter, Fullerton, California, USA).
Determination of activities of mitochondrial ETC complexes
COX (complex IV) activity was assayed in platelets according to the method of Sottocasa et al. 12 The assay mixture contained 0.3-mM reduced cytochrome c in 75-mM phosphate buffer (pH 7.5), and the reaction was started after adding 0.1 ml platelet sample and change in absorbance was recorded at 550 nm for 2 min. The results were expressed as nanomoles of cytochrome c oxidized. NADH dehydrogenase (complex I) activity was measured by the method of King and Howard. 13 The reaction mixture contained 0.2 M glycyl glycine buffer, pH 8.5, 6 mM NADH in 2 mM glycyl glycine buffer and 10.5 mM cytochrome c. To this, platelet sample (0.1 ml) was added to start the reaction, and the change in absorbance was noted at 550 nm for 2 min. The results were expressed as nanomoles of NADH oxidized. Succinate dehydrogenase (complex II) activity was measured as per the method of King. 14 The reaction mixture contained 0.2 M phosphate buffer pH 7.8, 1% bovine albumin, 0.6 M succinic acid and 0.03 M potassium ferricyanide. The reaction was initiated by the addition of platelet sample (150 µl) and change in absorbance was measured at 420 nm for 2 min. The results were expressed as nanomoles of succinate consumed.
Determination of activities of antioxidant enzymes
SOD activity was measured by the method of Asada and Kiso. 15 Mixture of platelet sample (0.1 ml), distilled water, ice-cold methanol and chloroform was centrifuged and the supernatant was used for the assay of SOD. Reaction mixture consisted of 100 mM sodium carbonate buffer (pH 10.0), 96 mM nitro blue tetrazolium and 0.6% Triton X-100. The reaction was initiated by the addition of 20 mM hydroxylamine hydrochloride, pH 6.0, and change in absorbance was measured at 560 nm for 3 min. The enzyme activity was expressed as units per milligram protein, where 1 unit of enzyme is defined as the amount of enzyme required to produce 50% inhibition.
The determination of CAT activity was carried out according to a modified method of Nelson and Kiesow. 16 The reaction was initiated by adding 0.07 ml of 0.3 M aqueous hydrogen peroxide solution to an aliquot 0.02 ml of platelet sample in potassium phosphate buffer (pH 7.0) and change in absorbance at 240 nm was measured for 2 min. The results were expressed as picomoles of hydrogen peroxide consumed.
GPX activity was measured by the method of Necheles et al. 17 Platelet sample of 50 µl was added to the reaction mixture consisting of 0.4 M phosphate buffer, pH 7.0, 10 mM sodium azide, 8 mM glutathione and hydrogen peroxide and incubated at 37°C for 3 min. A 10% trichloroacetic acid was then added and centrifuged; a supernatant of 0.5 ml was taken and 0.3 M disodium hydrogen phosphate followed by DTNB was added to it. Absorbance was recorded at 412 nm within 5 min, and the results were expressed as micrograms of glutathione consumed.
Determination of oxidative stress
The carbonylation of plasma proteins was determined by modified Levine et al.’s method. 18 From 0.5 ml plasma, the proteins were precipitated and treated with dinitrophenylhydrazine. It was then washed and dissolved in 1.5 ml of protein-dissolving solution (2 g sodium dodecyl sulfate and 50 mg EDTA in 100 ml of 80 mM phosphate buffer, pH 8.0). The color intensity of the supernatant was measured at 370 nm against 2 M hydrochloric acid. Lipid peroxidation in plasma was assayed by the method of Buege and Aust. 19 Plasma of 1.0 ml was mixed with 2 ml of mildly heated thiobarbituric acid and treated appropriately to remove the supernatant. The absorption was read at 535 nm against appropriate blank. Total blood thiols were assayed by the method of Ellman. 20 DTNB was added to the reaction mixture with whole blood (0.02 ml), and absorbance was determined at 420 nm after 1 h. The results were expressed as millimoles per liter.
Statistical analysis
The study had statistical power of 80%. The statistical analysis was undertaken using the SPSS v.16 software. All the values were expressed as mean ± SD. The normality and distribution of the data were checked by Q–Q plots and appropriate tests were applied as required. The serum parameters were compared only between ALP and healthy controls by student’s t test. Kruskall–Wallis test was applied to the other parameters to compare the three groups because they followed non-normal distribution. Mann–Whitney U test was used for pairwise comparison, and correlation was performed using Spearman’s correlation.
Results
Characteristics of patients
From February 2011 to November 2011, all the patients admitted with ALP poisoning were assessed, and of these, 21 patients were recruited as ALP group. All the cases were of suicidal ingestion and seven had received some treatment at local hospitals before reaching us. The mean dose of ALP ingested was 5.5 g (range 1.5–15 g), and the mean interval between ingestion and arrival to hospital was 3.88 h (range 0.67–8 h). The amount ingested was not known in three patients, whereas in four patients time of ingestion was not clear. Of the 21 patients, 13 (61%) died, with the majority of death occuring within 24 h of admission. Epigastric pain (90%) and vomiting (85%) were the most common symptoms. The clinical features of ALP group patients are shown in Table 1. Shock group consisted of 22 patients (men = 10; women = 12). Of these, 11 had cardiogenic shock, nine had septic shock and two had hemorrhagic shock. A total of 32 healthy adults (men = 17; women = 15) were included in the control group (Table 1).
Subject demographics and clinical features in ALP poisoning
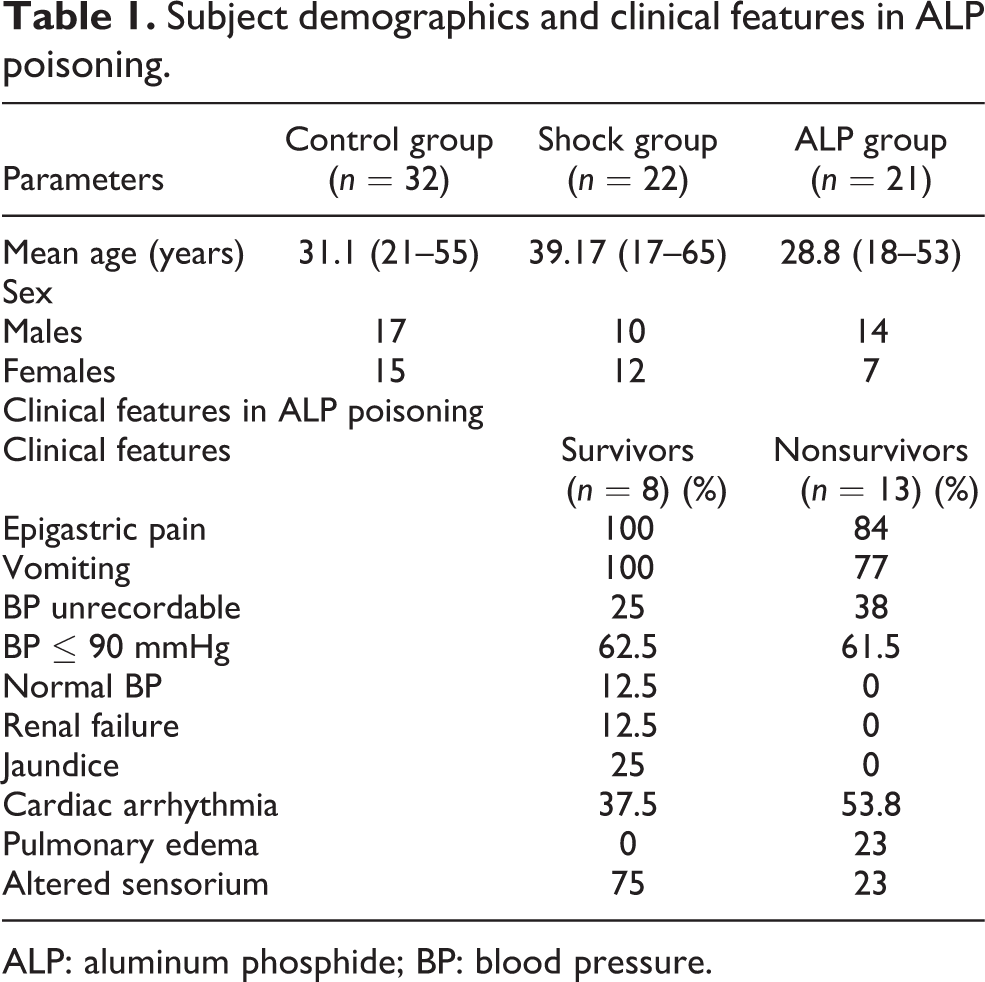
ALP: aluminum phosphide; BP: blood pressure.
Profile of serum biochemical parameters in ALP poisoning
In the serum, 20 basic biochemical parameters were assessed using Hitachi modular P800, and the means were compared with those of healthy controls using Student’s t test. Except for serum levels of creatine kinase-muscle brain (CKMB), lactate dehydrogenase, urea and creatinine, which were significantly elevated in the ALP group (p < 0.01), other parameters were within normal limits. CKMB was raised in almost all the patients suggesting myocardial injury, whereas there was only a mild but significant rise in both urea and creatinine, indicating renal injury possibly due to shock (Table 2).
Serum biochemical profile in ALP poisoninga
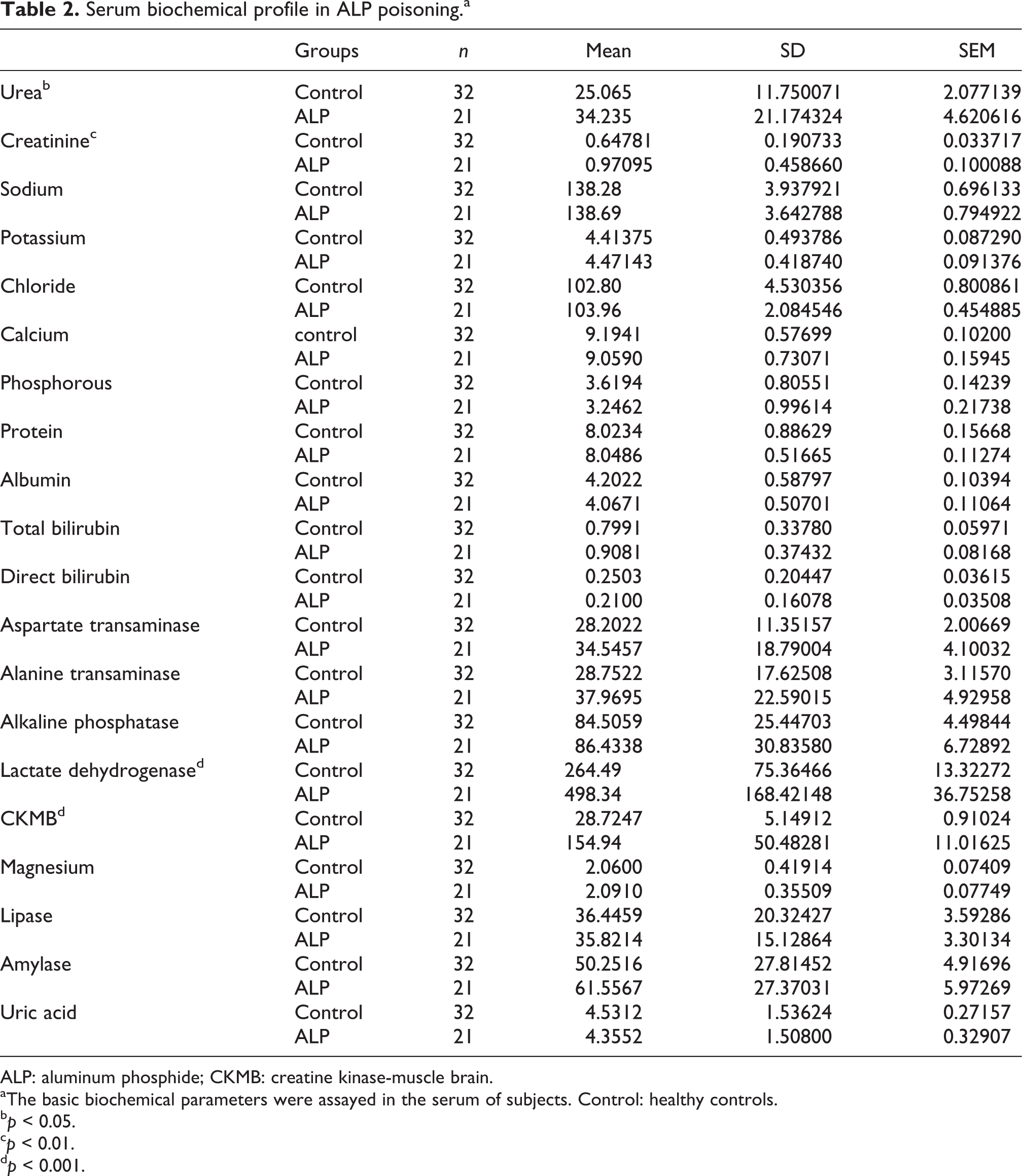
ALP: aluminum phosphide; CKMB: creatine kinase-muscle brain.
aThe basic biochemical parameters were assayed in the serum of subjects. Control: healthy controls.
b p < 0.05.
c p < 0.01.
d p < 0.001.
Activity of mitochondrial respiratory complexes
The COX (complex IV) activity was about 12% and 37% less in shock and ALP group (p < 0.001), respectively, when compared with controls. NADH dehydrogenase (complex I) activity was similar in both control and shock groups, whereas in the ALP group, the activity was lower by 45% (p < 0.001). Succinate dehydrogenase (complex II) activity in human platelets was not affected in controls and shock patients, whereas in ALP group, it was reduced by 31% (p < 0.001; Table 3).
Activities of mitochondrial complexes and antioxidant enzymes in ALP poisoninga
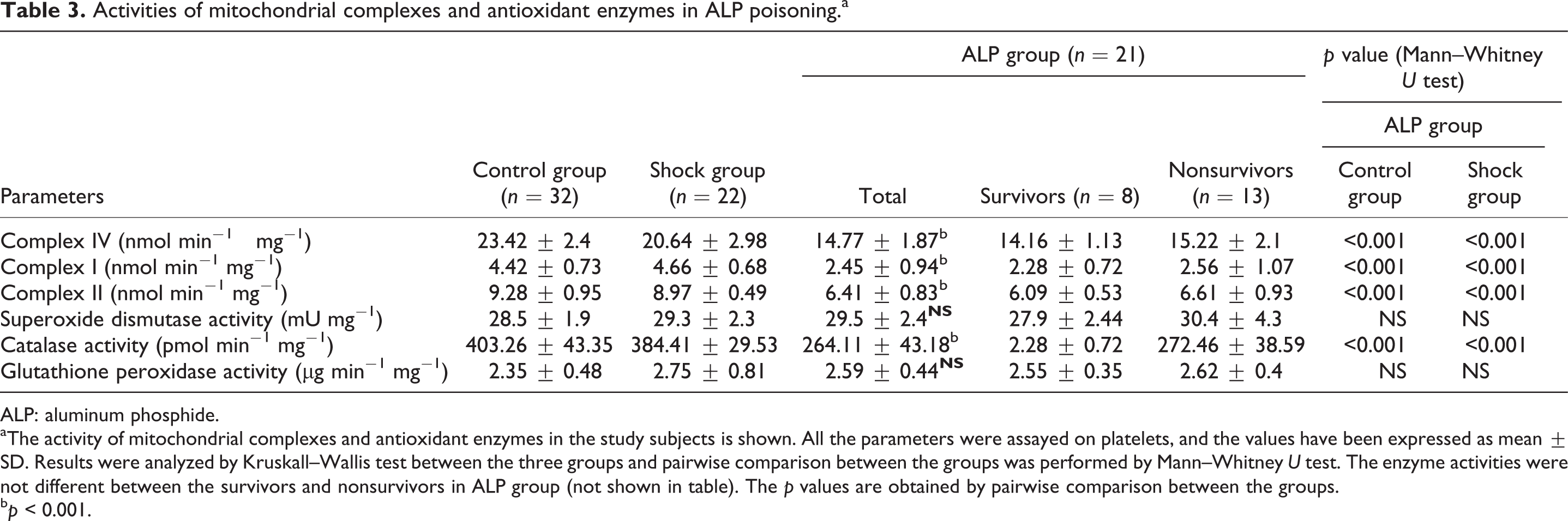
ALP: aluminum phosphide.
aThe activity of mitochondrial complexes and antioxidant enzymes in the study subjects is shown. All the parameters were assayed on platelets, and the values have been expressed as mean ± SD. Results were analyzed by Kruskall–Wallis test between the three groups and pairwise comparison between the groups was performed by Mann–Whitney U test. The enzyme activities were not different between the survivors and nonsurvivors in ALP group (not shown in table). The p values are obtained by pairwise comparison between the groups.
b p < 0.001.
Activity of antioxidant enzymes
The CAT activity was less by 35% in the ALP group compared with control and shock groups (p < 0.001). The GPX in platelets was found to be insignificantly higher in both shock and ALP groups. SOD activity was unaffected in both shock and ALP groups (Table 3).
Oxidative stress in ALP poisoning
The total blood thiol levels were significantly lower in both shock and ALP groups (p < 0.001) than in controls by 20% and 34%, respectively (Figure 1). There was a 31% increase in malonidialdehyde content in shock patients, whereas a nearly twofold increase was observed in patients with ALP poisoning (p < 0.001) when compared with controls. The levels of protein carbonylation are shown in Figure 1. It was 30% higher in shock patients, whereas in ALP patients, it was increased by 60% (p < 0.001).

Oxidative stress parameters in ALP poisoning. The markers of oxidative stress- lipid peroxidation, protein carbonylation and total blood thiol levels were estimated in the subjects. Lipid peroxidation and protein carbonyl content were assayed in plasma and total thiols in blood. The level of total thiols was significantly lower while plasma lipid peroxidation and protein carbonylation was significantly higher in ALP group. The values are expressed as mean ±SD and plotted against the X axis.
Others
The enzyme activities of complexes I, II and IV on linear regression analysis showed a significant dependence of complex IV activity on both complexes I and II (R 2 = 0.997). Significant statistical correlations were also observed between the activities of complexes I, II and IV as well as CAT in the ALP group. Spearman’s correlation was undertaken between the above biochemical parameters and survival status in ALP group; however, no significant correlation could be observed (data not shown, see supplementary data for more information).
Discussion
ALP ingestion with suicidal intent is common in countries like India and Iran and is associated with high mortality. Following ingestion, phosphine is released, which gets absorbed rapidly from the gut into the systemic circulation. The tissue distribution, mechanism of action and elimination of phosphine are still unclear, 21 although the manifestations become apparent within as early as 1–2 h of ingestion. The most common features associated with ALP ingestion are a garlic odor in breath, vomiting, epigastric pain, diarrhea and hypotension. The cause of death is refractory shock in most cases. 21
The exact mechanism of action of phosphine is not known and the available evidences are scattered across species. The exposure conditions of the toxin and different physiology between invertebrate and vertebrate organisms precludes a clear direct comparison of phosphine toxicology. Studies on invertebrates typically involve phosphine fumigation at low concentrations for a period of 20–48 h, simulating the situation when grain is fumigated. On the other hand, laboratory studies on rats or mammalian cell lines involve exposure to ALP as a solid formulation or in solution at high concentrations approximating the clinical situation of attempted suicide. Despite the obvious differences, there are some similarities in the responses that phosphine elicits and deserves attention. 21,22 The common features include an initial state of agitation/hyperactivity followed by lethargy, decrease in metabolic output and increase in oxidative stress. Late manifestations are anesthetization in insects and nematodes, whereas vertebrates exhibit pulmonary edema, inhibited oxygen transport, metabolic acidosis, hypotension and cardiac failure. 23
We investigated the effect of ALP on the activity of mitochondrial respiratory complexes in platelets and observed a decrease in the activities of complexes I and II along with complex IV. The activities of complexes I, II and IV were lesser by 45%, 31% and 37%, respectively, with inhibition being statistically significant for all the three complexes. The effect of ALP on COX in human platelets in severely poisoned patient’s with ALP has also been shown in an earlier study. 1 The current results from humans are similar to animal experiments, where acute ALP exposure produced a decrease in the activities of mitochondrial complexes I, II and IV in rat brain and liver. 6,7
Strong corroborative evidence suggests that phosphine disrupts mitochondrial energy metabolism in insects, nematodes and rats. Phosphine has been shown to inhibit respiration in intact nematodes, 24 mitochondria of rat liver 7,25 and insects. 4,26 A direct correlation has been observed between the degree of suppression and mitochondrial respiration. Living organisms and insect pests are not affected when exposed to phosphine under hypoxic conditions 27 ; conversely in the conditions of activated metabolism and increased metabolic demand (i.e. under hyperoxic conditions 28 and mitochondrial uncouplers 29 ), increased sensitivity toward phosphine is observed suggesting a relationship between phosphine toxicity and the rate of mitochondrial respiration. Further evidence from nematodes suggests COX to be the primary site of interaction with phosphine in the electron transport chain (ETC). 4 Phosphine also seems to alter the oxidation status, specifically, of cytochrome a of complex IV. 8 Evidences also show a decrease in mitochondrial oxygen consumption, adenosine triphosphate (ATP) levels, ATP synthesis and ATP hydrolysis 7 culminating in a metabolic and energy crisis. 24,30 Thus, phosphine-induced mitochondrial dysfunction is an important component of its toxicity.
Furthermore, following ALP exposure, we observed a decrease in the activity of CAT, but the activities of SOD and GPX were unaffected. Earlier studies on rat brain have also shown a decrease in the activities of CAT, SOD and glutathione reductase, while that of GPX was unaffected. 5 The effect of phosphine on antioxidant isozymes still needs further probing, however, we observe a similar pattern of CAT inhibition in both rats and humans. The plasma lipid peroxidation and carbonyl content were increased in both shock and ALP groups, with significantly higher levels in ALP group than shock group, while the total blood thiol levels were significantly lower. Phosphine-induced oxidative damage to macromolecules has already been demonstrated in insects, 31 nematodes, 29 mammalian cell lines 32 and rats. 33
The electron flow across the ETC, even under basal conditions, generates a small amount of superoxide through the inappropriate transfer of electrons to molecular oxygen, and when the flow of electrons is impeded, the production of superoxide can become very significant. 34 The decrease in mitochondrial complex activities and inhibition of CAT could compound the problem leading to a state of oxidative stress, and phosphine-induced generation of reactive oxygen species (ROS) could be a strong alternative model to mortality caused by energy insufficiency. The free radicals rapidly react with the neighboring molecules with resultant damage to lipids and proteins, in addition to depletion of reduced glutathione.
Hence, the pathophysiologic effects of phosphine toxicity could be the result of a combined energy insufficiency as well as oxidative stress, and hence a strategy to develop an effective management for phosphine toxicity must include both the mechanisms in mind. It also seems that phosphine does not have any target organ for damage but the organs that are mainly dependent on oxidative phosphorylation will be affected at the earliest, a possible reason for early cardiac dysfunction that occurs in ALP poisoning. The depletion of glutathione levels and evidence of free radical-induced damage in patients poisoned with ALP have prompted the use of N-acetylcysteine as a therapeutic measure to replenish the glutathione. N-Acetylcysteine was found to be beneficial in a study conducted in rats, in which the treatment improved the survival time 35 ; however, in a small study in humans, N-acetylcysteine was seen to offer no benefit (unpublished data). Many antioxidants have been tried to counter phosphine toxicity and compounds such as melatonin have shown good promising results in vitro. 33,36 However, concerns have also been raised regarding the impermeable nature of inner membrane of mitochondria, in which case, mitochondrial-specific antioxidant compounds such as Mito-Q may offer benefit. The occurrence of energy insufficiency in phosphine toxicity has been the rationale for trying agents such as trimetazidine, which switches myocyte metabolism to glucose from fatty acids, thus reducing oxygen consumption and may have a potential role. 37 Phosphine also inhibits cholinesterases, the significance of which is less understood. Interestingly, there are some instances where organophosphates have also been reported to impair mitochondrial function. 38,39 The muscarinic acetylcholine antagonist, atropine, and a cholinesterase reactivator, pralidoxime, have been shown to provide protection in rats against phosphine exposure indicating that acetylcholine signaling may also be an important component of phosphine toxicity. The underlying link if any between cholinesterase inhibition and mitochondrial function is yet to be determined.
Our study, however, has certain limitations which need consideration. The activities of mitochondrial respiratory complexes I, II and IV were studied individually, and parameters to assess the effect of ALP on ETC and oxidative phosphorylation (OXPHOS) as a whole, such as cellular oxygen consumption, were not studied. We also could not assess the cellular ATP levels in the groups; hence, although mitochondrial dysfunction is evident, proof of energy insufficiency in humans is still to be shown. We also could not study the effect of ALP on different antioxidant isozymes and the levels of ROS could not be measured directly. Thus, it is not clear that elevated markers of oxidative stress were due to the direct effect of ALP or secondary to the inhibition of CAT.
Conclusion
In humans, following ALP ingestion, not only COX but also other enzymes are affected leading to energy insufficiency and increased ROS production, which both synergistically can lead to tissue damage. The findings of the present study possibly direct to certain future implications in the management of patients poisoned with ALP. Assaying the activity of mitochondrial complexes maybe utilized as a biomarker in ALP poisoning for the diagnosis and follow up. An interesting review has suggested the role of mitochondrial protection strategies in mitochondrial dysfunction induced by sepsis, 40 a similar mitochondrial dysfunction that occurs in these patients. Experimental studies could be planned to find the usefulness of mitochondrial protection strategies in ALP poisoning.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Acknowledgments
The help and guidance offered by Drs S Anand, K Madhanraj, P Sathish and S Hemant in statistical analysis and sample procurement are acknowledged.
Conflict of Interest
The authors declared no conflicts of interest.