
Abstract
Cobalt exerts well-known and documented toxic effects on the thyroid, heart and the haematopoietic system, in addition to the occupational lung disease, allergic manifestations and a probably carcinogenic action. Cobalt neurotoxicity is reported in isolated cases, and it has never been systematically treated. Bilateral optic atrophy and retinopathy, bilateral nerve deafness and sensory-motor polyneuropathy have been described long ago as a result of chronic occupational exposure to cobal powder or during long-term treatment of anaemia with cobalt chloride. Recently, some patients with high levels of cobalt released from metal prosthesis have been referred as presenting with tinnitus, deafness, vertigo, visual changes, optic atrophy, tremor and peripheral neuropathy. The aim of this work is to group these cases and to identify a possible mechanism of cobalt neurotoxicity, focusing on hypothetic individual susceptibility such as altered metal-binding proteins, altered transport processes in target cells or polymorphic variation of genetic background.
Introduction
Exposure and effects of cobalt (Co) have been described in many studies, reviews, and toxicology books. Some important reviews1–6 showed that the main acute and chronic effects concern the respiratory system, heart and thyroid. A genotoxic and carcinogenic action was described in the International Agency for Research on Cancer (IARC) monograph7,8 in which Co is classified as group 2A (probably carcinogenic to humans).
Isolated case reports of neurological symptoms after exposure to high doses of Co were also described, following professional exposure, therapeutic ingestion of cobalt chloride (CoCl2) and metallic ion release from cobalt–chromium (Co-Cr) alloy in orthopaedic implants (prosthesis).9–19
A total of 11 case reports of neurological involvement due to heavy Co intoxication have been published in the last 60 years, 4 of them in the last 2 years, describing the effects of Co released from prostheses in human central nervous system.16–19 In April 2010 the United Kingdom’s Medical Products and Healthcare Devices Regulatory Agency published a medical device alert that recommended follow-up of metal-on-metal (MoM) hip replacement patients at least annually for 5 years postsurgery in order to reveal its toxicity symptoms. 20 However, this document does not take into consideration the effects of Co on the nervous system and the treatment options of Co neurotoxicity. Nevertheless, it seems accepted that there have been about 1 million MoM hips implanted worldwide in the past 15 years, which may be at risk of arthroprosthetic cobaltism. 21
The aims of this review are to describe the published evidences of Co neurotoxicity, to identify possible mechanisms that influence toxicity and to explain individual susceptibility to it.
Physical and chemical properties of Co
Co is a relatively rare element in the earth’s crust, which is essential to mammals in the form of cobalamin (vitamin B12). Pure Co metal is steel-grey, shiny, hard, ductile, brittle and magnetic with properties similar to those of iron and nickel. It is commonly found with copper, iron, lead, nickel and silver ores in concentrations less than 1%. Co occurs in the (0), (II) and (III) valence states; Co (II) is more stable than Co (III), which is a powerful oxidizing agent that can oxidize water and deliver oxygen. Metallic Co (0) occurs in two allotropic forms, hexagonal and cubic, which are stable at room temperature. 3
Sources of Co in the air are both natural (volcanic eruptions, erosion, forest fire, seawater spray) and anthropogenic (burning of fossil fuel, engine emissions, sewage sludge and processing of Co-containing alloys). Typically the mean concentration in air is small (about 1–2 ng/m3) in urban areas and less in remote areas. 22
The use of Co compounds for the manufacture of blue-coloured pottery and glass antedates the Christian era, but the main consumption of Co is now in the production of steel and alloys. Advantages of Co alloys are high melting point, strength and resistance to oxidation. The alloys are made of Co, Cr and other metals (tungsten but also molybdenum, tantalum, niobium, zirconium and hafnium) and are used to produce very hard cutting tools and surfaces subject to heavy wear (e.g. in turbines or space vehicles).5,6 Less common uses include catalysts in the chemical and oil-refining industry, drying agents in paints, electroplating, radiology and an additive in fertilizer and animal feed. 3
For the general population the diet is the main source of exposure to Co; human dietary intake of Co is highly variable, with reported values generally between 5 and 50 μg/day. A diffuse and increasing use of Co alloys in orthopaedic implants that require high resistance, such as knee and hip, has become another important route of exposure. In the occupational setting exposure to Co occurs primarily during Co powder and hard metal production and diamond polishing.23,24
The main biologically active Co compound is vitamin B12, or cyanocobalamin, in which Co is complexed with four pyrrole nuclei joined in a ring. Vitamin B12 acts as a coenzyme in many enzymatic processes and catalyzes reactions such as the synthesis of methionine, the metabolism of purines and folates and the formation of methylmalonic acid in succinic acid. 25 The normal plasmatic concentrations of Co and vitamin B12 are <0.2 ng/ml and 200–900 pg/ml, respectively. 5
Co neurotoxicity in occupational exposure
Inhalation of Co dust represents the main source of exposure; even in some experimental and clinical conditions the absorption through intact skin has also been demonstrated.26,27 Co-containing dust may be released into the workplace air during the production of Co compounds mainly during hard metal alloy processing. Characteristic workplace concentrations ranged from 0.01 to 1.7 mg/m3.24,28,29 In exposure assessment, urinary cobalt (CoU) is the most used biomarker and it enables to quantify the occupational exposure. However, should be considered that CoU rapidly increased in the hours following the cessation of exposure, with a peak of excretion about 3 hours after the end of exposure. 24
The American Conference of Governmental Industrial Hygienists (ACGIH) classifies Co as A3 (animal carcinogen) and established a Biological Exposure Index of 15 μg/L of Co in urine and 1 μg/L in blood at the end of workweek and this is equivalent of a TLV-TWA of 0.02 mg/m3. 30 For Co-U, the Deutsche Research Foundation (DFG) fixed the exposure equivalents for carcinogenic materials at 6, 15, 30, 60 and 300 μg/L for airborne Co concentrations (Co-air) of 10, 25, 50, 100 and 500 μg/m3. 31 The representative Co concentrations in serum and urine of occupationally exposed workers are reported in Table 1 .
Levels of cobalt in serum, whole blood and urine in workers exposed to cobalt
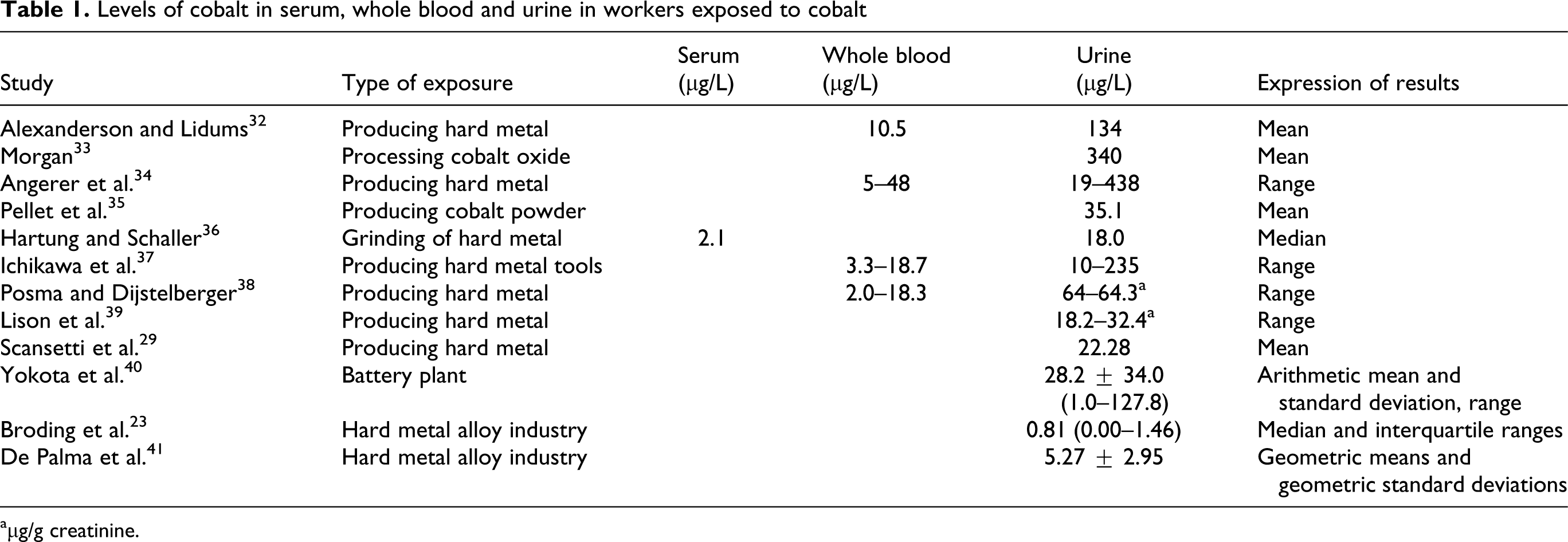
aμg/g creatinine.
In non-occupationally exposed subjects, the normal concentrations are 0.1–1.5 μg/L in urine, 0.05–1.5 μg/L in blood and 0.05–0.3 μg/L in plasma. Table 2 reports the reference values of Co in urine for the US population from the National Health and Nutrition Survey. 42
Geometric mean and selected percentiles of urinary cobalt concentration (µg/L) for the US population from the National Health and Nutrition Survey42

In exposed workers, in addition to respiratory and cardiovascular diseases and contact dermatitis, 43 some effects on the nervous system have also been reported, particularly memory loss, neuropathies and a decreased visual acuity12,13 (Table 3 ).
Cases of neurotoxicity reported in occupational exposures to cobalt

CoB: cobalt in blood.
Jordan and colleagues 12 reported significantly impaired attention (p < 0.05) and verbal memory loss (p < 0.001) in 12 hard metal workers exposed to tungsten carbide and Co (as dust and dissolved in an organic solvent) compared to 26 healthy unexposed controls. Memory deficits were related to difficulties in attention and verbal memory (measured using the Wechsler Memory Scale-Revised), with an apparent sparing of visual-spatial memory. All the exposed workers had pulmonary manifestations of disease and all of them complained of memory difficulties.
A 48-year-old worker exposed to Co dust for 20 months (50 hours a week) developed bilateral optic atrophy and bilateral nerve deafness. On examination his visual acuity was 6/36 in the right eye and 6/60 in the left eye, the temporal margins of the discs were pale and hearing was impaired bilaterally. Co concentration in blood was 234 μg/L and in urine 119 μg/24 h. Fourteen months after discontinuing work, his visual activity improved and hearing returned to normal. The definite improvement in his vision and hearing occurring within 1 month of discontinuing work, strongly suggests that Co toxicity was the cause of the deafness and visual loss. 13
Iatrogenic Co neurotoxicity
In the past, CoCl2 has been used in the treatment of refractory anaemia, nephritis and infections. Today this therapy is not used because of the rarity of positive responses and the frequency of adverse effects (mainly goitre, myxoedema and congestive heart failure). 1
Between 1950 and 1970 three cases of neurotoxicity following CoCl2 consumption were reported (Table 4 )
Neurotoxic effects related to cobalt consumption for anaemia treatment

Gardner 9 studied 17 patients with anaemia and uraemia treated with CoCl2, 4 patients noted tinnitus after 4–16 weeks of therapy and 1 developed nerve deafness after 12 weeks. Hearing was absent in a frequency range above 1000 cycles/s; within 10 weeks of stopping the drug, the hearing deficit recovered to the pretreatment range; in this patient nerve deafness recurred on re-starting therapy and recovered again when the drug was discontinued. The central nervous system was severely affected; the vestibular and cochlear parts of the eight nerves were damaged, as shown by the abnormal caloric tests and audiometric curves. Paraesthesia in the limbs, absent tendon jerks and calf tenderness indicated that the patient had a peripheral neuritis. The difficulty in walking was probably due to a combination of vestibular damage and peripheral neuropathy. A patient who received 400 mg of CoCl2 daily for 6 weeks complained of paraesthesias, burning sensation on the soles with numbness of the toes. Symptoms disappeared when the therapy was stopped.
After a 6-month treatment with CoCl2 (25 mg of CoCl2 daily), a 35-year-old woman developed limb paraesthesia, unsteady gait, impaired hearing and dizzy spells in addition to nausea, vomiting and weight loss. 10 Clinical examination confirmed bilateral nerve deafness, absent ankle reflexes and impaired vibration sense which all resolved within 4 months of Co withdrawal. Audiometry demonstrated a general decrease in sound perception of all frequencies, and caloric tests were also found to be abnormal. A Diagnosis of chronic renal failure complicated by CoCl2 poisoning was made; the combination of prolonged exposure to the drug and poor renal clearance probably represents the reason of the severe toxic effects found in this patient.
Licht and co-workers 11 reported a case of severe visual disturbances in a man treated with CoCl2 (200 mg daily) for pancytopenia and hypercellular bone marrow. An ophthalmological examination revealed bilateral reduced visual acuity (right eye 6/60; left eye 6/21). Fundus oculi examination revealed marked pallor of the right optic disk mainly on the temporal side and moderate temporal pallor of the left optic disk, suggesting both optic atrophy and abnormal choroidal perfusion. The peripheral visual field of the right eye showed a concentric constriction, more accentuated on the nasal side, and a central and paramacular scotoma. Both the peripheral and central visual fields of the left eye were normal.
The patient had received a total dose of 73 g of CoCl2 over a period of 3 years and almost half of this amount had been administered during the 4 months preceding the appearance of the visual disturbances. Following cessation of the drug, there was no further deterioration in vision despite the progression of the underlying disease. The assumption that the visual changes were due to a toxic effect of Co therapy is supported by the fact that there was no further aggravation in visual function following cessation of this therapy.
In these studies, there are no indications of Co body burdens, but the constant intake of CoCl2 for long periods leads us to consider that Co levels were very high.
Co neurotoxicity and prostheses
Release of Co ions from prosthetic materials occurs by wear, corrosion and mechanisms such as fretting, stress and fatigue.
Several studies have determined Co concentrations following hip replacement and during follow-up after implantation, many of which have compared implanted subjects with a control group. The release of metal ions is a well-documented consequence of total hip arthroplasty implants.44,45 In some of these studies, Co concentrations in subjects with prosthesis were statistically higher than those in the control groups (Table 5 ).
Study that have investigated the release of cobalt from metal-on-metal surface arthroplasty. The concentrations of cobalt measured in serum and/or whole blood are expressed in µg/L. In the column ‘Control group’, we reported the statistic results of the comparison between subjects with prostheses and controls

d: days, m: months, w: weeks, y: years, SD: standard deviation, no sign: nonsignificant, MoM: metal-on-metal, THR: total hip replacement, THA: total hip arthroplasty, ASR: surface replacement implants, PE: polyethylene, Co-Cr: cobalt–chromium, Ti: titanium, Co-Cr-Mo: cobalt–chromium-molybdenum.
High values of Co in biological matrices have been reported in six case reports.15–19,64 In these subjects, an unusually high amount of metallic debris were evident around the articulating space of the prosthesis (metallosis). However, it is interesting to note that in some studies there were the subjects “outsiders” with high levels of Co in serum or blood not evident in cumulative concentration expressed as geometric/arithmetic mean or median (Table 6 ).
The highest outlier of cobalt concentrations (µg/L) extrapolated from studies of subjects with orthopaedic implants

Follow-up studies reported a running-in phase during the first 2 years after surgery (0.5–2 million cycles) during which Co ion concentration is significantly elevated. 63 After this period, there is a steady state in which the ion concentration is constant until the implant begins to fail and ion concentration begins to rise again.65,66 A higher degree of running-in wear (15–20 μm/year) was quantified during the first year after surgery and a reduction to 2–5 μm/year after the second and third year. 67 More detailed investigations about prosthesis corrosion showed no statistically significative differences in Co concentration in the subgroup of different gender, age, body mass index and sports.54,55
Although Co endogenous exposure has been consistently documented in clinical series and mainly associated with metal ion release from metal prosthesis,68–70 neurotoxicity has been rarely described.
All the above-mentioned case reports,15–19 with the exception of Von Schewelov’s study, 64 reported neurological symptoms (Table 7 ).
Neurotoxic effects in subjects with prostheses and related cobalt levels
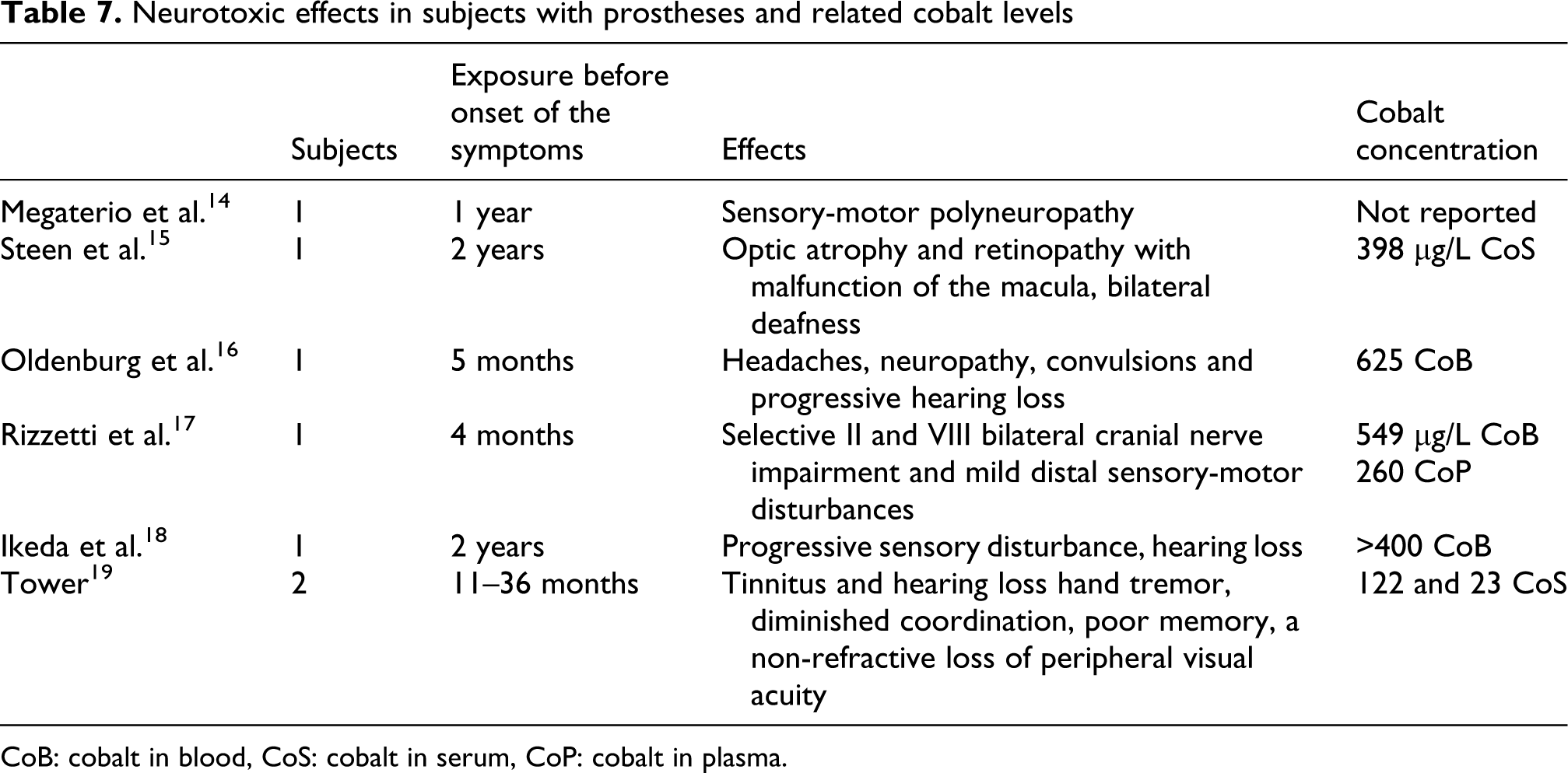
CoB: cobalt in blood, CoS: cobalt in serum, CoP: cobalt in plasma.
Megaterio and colleagues 14 described the case of a 47-year-old man who underwent revision of his right hip arthroplasty; 1 year after surgery, he developed thyreopathy and sensory-motor polyneuropathy. The next year, hip arthroplasty was removed and the Co and Cr concentrations in urine and blood, were higher than the normal values.
Steens and colleagues 15 described a case of severe Co poisoning with neurological involvement after hip arthroplasty revision. A 53-year-old man, treated with a cemented total hip arthroplasty including a ceramic-on-ceramic pairing 6 years previously, underwent revision surgery 3 years later because of hip pain. Two years after surgery, he developed visual loss (he could just recognize outlines and colours but could not read) and deafness, in addition to numbness in his feet. Because of pain in his hip, radiographs performed, which showed prosthesis deterioration and wear. A massive metallosis and more than 500 ml of metallic black synovial fluid were found during revision, and examination of cerebrospinal fluid also showed an elevated Co concentration (3.2 μg/L). An ophthalmological examination revealed toxic atrophy of the optical nerve and retinopathy with malfunction of the macula. After prosthesis removal the hearing loss recovered and the numbness of the feet disappeared while vision recovered only partially.
Three years later, Oldenburg and co-workers 16 reported the case of a 55-year-old man who experienced a massive deterioration of his metal femoral head by overlooked particles of the previously broken ceramic head; Co concentration in biological fluids was severely higher than normal value, and he displayed a severe Co intoxication with multiorganic disorder consisting of hypothyroidism, cardiomyopathy and the involvement of the central nervous system with headaches, neuropathy, convulsions and progressive hearing loss. Moreover, an increased distal sensory latency and a decreased conduction velocity of nervus peroneus were found. The echocardiogram demonstrated a moderate reduced systolic left ventricular function with concentric left ventricular hypertrophy, and the myocardial biopsy showed interstitial fibrosis. The authors described that 6 months after the revision surgery the symptoms subsided completely however in the long-term follow-up the patient’s neurologic symptoms have persisted but without describe it.
Tower 19 reported two cases of Co neurotoxicity in hip replacement. A 49-year-old male reported anxiety, headaches, tinnitus and hearing loss 18 months after the implant; hand tremor, diminished coordination, slow cognition and poor memory after 30 months and a non-refractive loss of peripheral visual acuity after 36 months. Diasystolic dysfunction was found on interval echocardiography. Laboratory findings included a serum Co level of 83 μg/L, a cerebrospinal fluid Co level of 2.2 μg/L and a joint fluid Co level of 3200 μg/L. Eleven months after revision, the patient’s hip pain, affect, cognition, hearing, exercise tolerance, tremor and professional productivity improved, while his tinnitus and visual symptoms were stable.
The second case was about a 49-year-old male who complained of mental fog, memory loss, vertigo, hearing loss and dyspnoea 11 months after the metal on metal hip arthroplasty.
In both cases, the periprosthetic tissues showed necrosis, staining with metal debris and visible wear of the retrieved bearing. All symptoms, except the visual changes in the first case, improved 3 and 6 months post revision.
Recently, Ikeda and colleagues 18 presented the case of a 56-year-old woman with a detailed analysis of polyneuropathy associated with artificial joint-related metallosis. She developed progressive sensory disturbance, hearing loss and hypothyroidism. Neurologically, she had painful dysaesthesia and distal paraesthesia in the elbows and knees, and her joint position sense was impaired in the fingers and feet. She had distal dominant slight muscle weakness, no deep tendon reflexes and bilateral sensorineural hearing loss. Sural nerve biopsy indicated axonopathy; nerve conduction studies showed that no sensory nerve action potentials could be evoked, but the results of motor conduction studies were normal. Pure tone audiometry showed hearing thresholds of 52 and 45 dB in the right and left ear, respectively. In this case, the replacement of ceramic liner fractured with new bearing surface that had a Co-Cr alloy head and a polyethylene liner may be the cause of metallosis observed, the ceramic fragments in the joint space can damage the new metal prosthesis forming metal debris. After revision surgery, blood levels of cobalt decreased, and her symptoms improved.
Two years ago, we reported the case of a 58-year-old woman presenting high concentration of Co released from a hip arthroplasty with hypothyroidism, a selective II and VIII bilateral cranial nerve impairment and mild distal sensory-motor disturbances (numbness and weakness in her lower limbs). Increased concentrations of Co and Cr were found in blood, plasma, urine and cerebrospinal fluid (whole blood: 549, plasma: 260, urine: 1100 and cerebrospinal fluid: 11.4 μg/L). Laboratory investigations ruled out haematologic, infectious, neoplastic, metabolic and immunologic diseases. Concomitantly, the patient underwent various instrumental investigations including brain MRI (revealing hyperintensity of optic nerves and traits), electromyography nerve conduction (EMG/NC; showing mild lower limbs nerve amplitude reduction), acoustic and visual evoked potentials (positive for bilateral absence of brain stem acoustic responses and irregular cortical visual responses). Some months later, she became definitely blind, severely deaf and wheelchair bounded due to severe limb motor weakness. The decreasing Co levels after chelation therapy and removal of the implant gave rise to an improvement in symptoms, especially acoustic deficits. During prosthesis removal a massive infiltration of periprosthetic tissues by metallic debris became evident; subsequent analysis of these fluids displayed elevated concentrations of Co and Cr (404 and 285 mg/L, respectively). In the following 8 months, the patient showed a progressive neurologic improvement leading to the functional recovery of hearing and sensory-motor function with only a partial increase in sight, in spite of the metal levels definitely decreased but still far above the reference values. 17
In all the case reports, except the two cases by Tower, 19 summarized above patients underwent revision surgery, and metal-on-polyethylene components were used after the fracture of a ceramic head. After this kind of revision, extraordinary severe metallosis were observed by several authors and some of them concluded that the use of metal-on-polyethylene articular pairing is contraindicated after ceramic head fracture65,71; in fact fine ceramic particles can remain in situ and can cause the destruction of all kinds of metals, by acting like a grinding wheel. 72
In 2010, the United Kingdom’s Medical Products and Healthcare Devices Regulatory Agency published a medical device alert that recommended following the MoM implantees at least annually for 5 years postsurgery. The alert also recommended to measure Co and Cr in blood and to carry out imaging studies for patients who report painful hip replacements.
In order to identify patients who require closer surveillance, a follow-up determination 3 months after the first test is recommended in patients with Co or Cr >7 μg/L. 20 It is interesting to note that the proposed level of 7 μg/L is higher than the Biological Exposure Index of ACGIH for occupationally exposed people (1 μg/L Co in blood). Most patients implanted with MoM bearings have Co levels greater than those allowed in exposed workers and consequently they may develop an increased incidence of subclinical cognitive and cardiac impairments.
All cases, except the second reported by Tower 19 where serum the Co concentration was 23 μl (but Co level in joint fluid was 3300 μg/L), were characterized by high concentrations of Co. In the case reported by Von Schewelov and colleagues, 64 a woman with metallosis and high levels of Co (92 μg/L) 4 years after a hip resurfacing procedure with a MoM hip implant had no symptoms except mild discomfort and instability in her hip. Only Oldenburg et al. 16 and Tower 19 reported cardiac manifestations similar to the symptoms reported after occupational and iatrogenic exposures. 6
A determinant factor in the onset of neurological symptoms is certainly the massive amounts of Co for a long period, even though probably there are individual and predisposing factors that explain the low incidence of neurotoxicity among all the subjects with metal prosthesis.
Co toxicokinetics
Co distribution can be influenced, as other metallic elements, by binding to the plasma proteins. It has been reported that albumin and transferrin are the major serum protein carriers for Co in the blood. Co is transported in circulation by albumin, 73 and several studies indicated that the metal is bound to the N-terminal peptides of albumin at the sequence Asp-Ala-His-Lys, similar to copper and nickel.74–76 However, a recent study demonstrated that Co also binds to sites A and B on the albumin molecule. 77
The Co binding to albumin has been studied because it is the basis of the so-called Albumin Cobalt Binding test (ACB test) approved by the Food and Drug Administration for myocardial ischaemia evaluation. The alteration of the N-terminus of human serum albumin gives rise to the commonly defined ischaemia-modified albumin (IMA).75,78,79 However, other pathologies such as trauma, scleroderma, diabetes, bacterial or viral infections, end-stage renal disease, liver cirrhosis, brain ischaemia, peripheral arterial disease and cancer are able to influence albumin binding to Co.80–82 Co binding to human serum albumin is also reduced in the presence of copper; Bar-Or et al. 76 showed that addition of equimolar concentrations of copper before Co addition in normal plasma resulted in no Co-albumin species formation. In addition, there are also evidences of possible genetic variants of human serum albumin, in particular structural changes at the N-terminus abolish transition metal binding.83,84
Co ions can also bind to transferrin, a family of large non-haeme iron-binding glycoproteins with a molecular weight of 80 kDa involved in the iron uptake. 83 To determine the stability of Co-transferrin complex in the presence of albumin, Smith 84 incubated a complex with albumin and showed that the absorbance of the complex decreased by about 50%, suggesting that half of the Co from Co-transferrin had been removed. Moshtaghie and co-workers 85 in a study of Co and iron binding to human serum transferrin showed that iron binding was reduced by 20% when 225 nmol/ml of Co was added to the reaction mixture.
For these reasons, it might be useful to measure the Co in the various blood fractions to assess the distribution of the metal and to quantify the binding with the plasmatic proteins.
The uptake of Co by the cells represents another important step. Even if it is not known whether Co enters mammalian cells via a specific transporter, it has been shown that P2X7 transporter is involved in the uptake of divalent cations and Co. 86 In the same way, the divalent metal transporter 1 (DMT1) previously known as DCT1, NRAMP2 or Slc11a2 is a active transporter for iron, zinc, manganese, Co, cadmium, copper, nickel and lead.87,88
DMT1 is identified as a key protein in the neuron defence mechanism against metal toxicity and it is regulated by a family interacting protein 1 (Ndfip1); in fact the induction of Ndfip1 expression protects neurons from metal toxicity while the absence leads to metal hypersensitivity. 89 We also mentioned the Zip family protein (ZRT- and IRT-like), a group of proteins with the capacity to transport different divalent cations into the cell. 90
Mechanism of action
The available data in scientific literature indicate that Co is cytotoxic to many cell types, including neural cells91–93 and it can induce cell death by apoptosis and necrosis. 94 Co can cause DNA fragmentation,95–97 activation of caspases, 94 increased production of reactive oxygen species (ROS),92,95,98 augmented phosphorylation of mitogen-activated protein (MAP) kinases93,94 and transcriptional repression of the human p53 gene. 98 Studies on the neurotoxic effects of Co in rats have shown behavioural alterations and depletion of neurotransmitters like dopamine, norepinephrine and serotonin.99,100 Co decreases postsynaptic responses induced by neurotransmitters in vitro and inhibits the synaptic transmission through a presynaptic blockage of calcium channels. 101
In an old experimental study, Alagna and D’Acquino 102 found pathological changes in the lens, retina, choroid and optic nerve following subcutaneous administration of CoCl2 in 16 rabbits (900 mg in 30 days). Fatty degeneration was found in the cells of the outer and inner retinal molecular layers and in the ganglion cells. Marked oedema, pigmentary disturbances and degenerative cellular changes were found in the choroid mainly in the posterior pole. The fibres of the optic nerves were swollen and tortuous with fragmentation in their myelin sheaths.
In 1994, a Polish study 103 reported atrophy of nerve fibres, injury to the ganglion, lesion in amacrine, bipolar and horizontal cells and alteration in the nucleus of photoreceptors in the retina of rabbits exposed intraperitoneally to CoCl2 (8 mg/kg/die for 25 days).
The Co action on the retina and optic nerve may have some analogy with the ophthalmic findings found in the toxic optic neuropathy due to other toxicants such as quinine, lead, tobacco and methanol.104–106
The toxic optic neuropathies are therefore ascribed to mitochondrial oxidative phosphorylation, similar from a clinical point of view to the congenital mitochondrial optic neuropathies, for example, Leber’s hereditary optic neuropathy and Kjer’s optic neuropathy. It is known that many optic neuropathies of both genetic and environmental origins, are determined by mitochondrial dysfunctions. 107
The mitochondria appeared to be a principal target of Co toxicity, as shown by the loss of mitochondrial membrane potential and the apoptogenic factors release from mitochondria. 108 At normoxic conditions, Co is able to stabilize the α-subunit of hypoxia-inducible factor (HIF-1α) by blocking its ubiquitination and proteasomal degradation. Increased levels of HIF-1α result in higher transcription of a set of genes that encode several proteins, for example, glycolytic enzymes, erythropoietin and heat shock proteins, important for the adaptation of cells to hypoxic stress.109,110 Recent data support an important role of HIF-1α in modulating mitochondrial function. Two studies described HIF-1-dependent induction of pyruvate dehydrogenase kinase-1, which reduces mitochondrial oxygen consumption and ROS production during hypoxia.111,112 In addition, HIF-1 alters electron transport chain function by mediating switching of a subunit of complex IV. 113
The effects of Co on auditory functions can also be associated with a toxic neuropathy. The ototoxicity of some drugs (antibiotic, anti-malarial and antineoplastic) is caused by the production and action of ROS on basal outer hair cells that appear to be more sensitive. 114
It has been shown that basal hair cells of guinea pigs contain lower amounts of the free radical scavenger glutathione and that the addition of this agent, and/or other antioxidants, influences the susceptibility gradient to neomycin. 115 Other mechanisms such as aminoglycoside-induced hair cell death are not so clear, even if it is estimated that 17–33% of cases are associated with mutations of the mitochondrial ribosomal RNA (rRNA) gene. 116 An analogue Co ototoxicity occurs with cisplatin that generates ROS in explants of cochlear tissues. 117 ROS can deplete cochlear tissues of antioxidant protective molecules, for example, glutathione and antioxidant enzymes; this may allow lipid peroxidation, as shown by increased concentrations of malondialdehyde, toxic lipid peroxides and aldehydes (e.g. 4-hydroxynonenal and peroxynitrite).118,119
It is known that Co ions are able to induce the ROS formation in vivo and in vitro. Co catalyzes the generation of hydroxyl radicals from hydrogen peroxide in a Fenton type reaction. The generation of ROS during hypoxia as well as in the presence of Co has been described.120,121 Exposure to Co can result in the rise of oxidative stress, decrease of reduced glutathione levels and the gain of oxidized glutathione levels, in addition to activation of the hexose monophosphate shunt and damage to DNA free-radical induced.121–125
A preferential damage to cochlear hair cells may be explained by heteroplasmic mutations occurring in the mitochondria of specific organs, such as in Leber’s hereditary optic neuropathy. Genetic predisposition to hearing loss induced by cisplatin may be related to mitochondrial mutations. Among 20 patients with ototoxic effect by cisplatin, 5 were found to cluster in a rare European J mitochondrial haplogroup that has been associated with Leber’s hereditary optic atrophy. 126 Ghelli and colleagues 127 showed that mitochondrial DNA haplogroups modulated the response of Leber’s optic neuropathy to 2,5-hexanedione, in particular, haplogroup J makes cells more sensitive to its toxic effect.
Furthermore, even the OPA1 gene is involved in autosomal-dominant optic atrophy that affects retinal ganglion cells and the axons forming the optic nerve, leading to progressive visual loss. Specific OPA1 mutations are responsible for other several distinct clinical presentations, such as dominant optic atrophy with deafness and severe multi-systemic syndromes which involve neurological and neuromuscular symptoms similar to those due to mitochondrial oxidative phosphorylation defects or mitochondrial DNA instability. 128
Finally, the findings brain MRI 17 in one patient with arthroprosthetic cobaltism and sural nerve biopsy in another 18 suggest that neurotoxicity occurs by demyelination and axonal loss. The role of ROS in demyelination is demonstrated and the mechanism of action known.129,130 Oxidative stress can damage the lipids, proteins and nucleic acids of cells and mitochondria. Oligodendrocytes are sensitive to oxidative stress in vitro, seemingly due to a diminished capacity for antioxidant defence and the presence of raised risk factors. Oxidative stress might therefore result in selective oligodendrocyte death and thereby demyelination in vivo. The reactive species may also damage the myelin sheath, promoting its attack by macrophages. Damage can occur directly by lipid peroxidation and indirectly by the activation of proteases and phospholipase A2. 129
Conclusion
Bilateral optic atrophy, retinopathy, bilateral nerve deafness and sensory-motor polyneuropathy are sporadically reported in different situations characterized by Co exposures.
At present, the most frequent route of exposure is the release from metallic prosthesis, while in the past occupational or iatrogenic exposures were often signaled. If we consider that in the past 15 years there have been about 1 million of MoM hips implanted worldwide, this argument becomes timely and relevant.
Often, orthopaedic surgeons and the general medical community are unaware that subjects with high Co concentrations are at risk of progressive audio-vestibular, visual and cognitive impairment, and it is not yet known whether these impairments are completely reversible.
It is also possible that a subtle neurotoxicity, like minor alterations in hearing, sight, cognition and memory, in patients with MoM hip implants might be confused with the normal aging.
On the other hand, the small number of cases, with respect to the many people potentially exposed to Co, could be due to different contributory factors such as a random breakage of the prosthesis, an individual predisposition, altered metal-binding proteins, an alteration of transport processes in target cellular or a genetic susceptibility to develop the damage. The possibility of an individual susceptibility may be confirmed by the presence of subjects with high blood concentrations of Co but no clinical symptoms.
In our view, the mechanisms of binding and transport represent the key point for further considerations. The binding proteins besides influencing the proportion of free and active metal can interfere with the prosthesis interface. In fact once the Co is released from the prosthesis, its transport, absorption, excretion and deposition in tissues are strongly dependent on the type of Co biological binders. The association of the released metal to proteins, for instance, might increase the corrosion rate at the implant/biology interface by increasing the dissolution of the surface passive (oxide) film formed on metallic materials. However, an alteration of binding capacity can be easily detected by the determination of Co in different blood fractions.
The possible mechanisms of action include Co interaction with the mitochondria and the ROS formation with consequent probable mitochondrial neuropathies. It is well known that mitochondrial dysfunction may be caused by specific mutations in the mitochondrial DNA and may enhance the influence of some environmental factors. This predisposition is likely to be genetically determined with a possible role played by polymorphic variation of mitochondrial genetic background.
An interesting starting point for further investigations could be an experimental model of very high Co concentration exposure, that bypassing any subjectivity or individual susceptibility that influences the accumulation, could probably allow the observation of a possible neurotoxic effect.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.