
Abstract
Novel thermoplastic vulcanizates (TPVs) based on a coordination-crosslinking system composed of ethylene-vinyl acetate copolymer (EVA), nitrile butadiene rubber (NBR) and liquid nitrile butadiene rubber (LNBR) were prepared via dynamic vulcanization, and a simple and efficient strategy for fabricating thermally responsive shape-memory polymers (TSMPs) was developed. After etching, both EVA/NBR/LNBR TPV and EVA/NBR TPV exhibited distinct sea-island surface microstructures. The incorporation of LNBR into NBR effectively reduced the particle size of the cross-linked rubber phase in EVA/NBR/LNBR TPV, significantly enhancing the mechanical strength of the TPV. EVA, serving as the continuous phase, mainly contributed to the shape fixity capability of the dynamic vulcanizate system. In contrast, the highly elastic NBR/LNBR rubber particles acted as the dispersed phase, providing the primary driving force during shape recovery. The introduction of LNBR into NBR resulted in a slight decrease in the shape-fixity ratio (SF%) of EVA/NBR/LNBR TPVs, but led to a remarkable increase in shape-recovery ratio (SR%). By adjusting the mass ratio of EVA to the NBR/LNBR blend, the shape fixity and shape recovery behaviors of the EVA/NBR/LNBR TPV can be effectively controlled under thermal stimulation. Increasing the EVA content in the TPV improved the SF% but significantly reduced the SR%. The shape-memory performance of EVA/NBR/LNBR TPVs was strongly temperature-dependent. When both the fixing and recovery temperatures were set to 85°C, the EVA/NBR/LNBR TPV with a mass ratio of 80/20 exhibited SF% and SR% values above 94.0%, with a shape-recovery time of 15∼20 s, demonstrating excellent shape-memory behavior.
Keywords
Introduction
Over the past decade, rubber systems crosslinked through noncovalent interactions have gradually become a significant focus of research.1,2 However, most noncovalent interactions-such as ionic bonding and hydrogen bonding-exhibit relatively weak cross-linking strength, making them insufficient for demanding engineering applications. In contrast, coordination bonds, as one of the strongest types of noncovalent interactions, are capable of forming cross-linked networks that combine reversibility with structural stability. Consequently, coordination-crosslinked rubber systems have emerged as an essential research direction.3,4
Research on liquid rubbers dates back to 1944, when Hardman degraded solid rubber into a low-viscosity rubber capable of flowing at room temperature, thereby opening a new avenue for the rubber industry. 5 Liquid rubbers containing functional groups possess reactive sites that enable efficient chain extension and cross-linking, facilitating the formation of high-performance thermoset vulcanized rubbers. In some cases, three-dimensional network structures can even be rapidly established at ambient temperatures, resulting in significant improvements in vulcanizate properties.6–8 Liquid rubbers can be utilized in various forms: they can be directly processed into rubber products or employed as modifiers for thermosetting resins and other polymeric materials.9–11
Compared with most synthetic rubbers, nitrile butadiene rubber (NBR), as a highly polar elastomer, exhibits excellent oil resistance, abrasion resistance, and chemical resistance. Consequently, NBR has been widely used in various fields, including automotive manufacturing and aerospace. The abundant cyano groups (-CN) in NBR can provide high‐strength crosslinking through metal–polymer coordination interactions within the macromolecular network.12–14
Liquid nitrile butadiene rubber (LNBR) is an effective toughening modifier that can co-crosslink within the NBR network. Its incorporation reduces the migration of additives within the rubber and significantly improves their dispersion uniformity, thereby enhancing the performance of the resulting vulcanizates. 15 Compared with NBR, LNBR possesses a lower molecular weight while maintaining good compatibility with the NBR matrix. Introducing LNBR into NBR increases the intermolecular spacing, reduces molecular entanglement, and promotes chain‐segment mobility.16,17
Ethylene-vinyl acetate copolymer (EVA) is a typical semi-crystalline thermoplastic resin composed of polar vinyl acetate units and non-polar ethylene units. 18 Owing to its excellent processability, tunable mechanical behavior, and outstanding resistance to environmental stress cracking, EVA has been widely applied in wire and cable insulation, packaging films, foam materials, and hot-melt adhesives.19–21
Thermoplastic elastomers (TPEs) are a class of novel polymeric materials that exhibit elastic behavior at room temperature while being capable of plastic deformation and melt processing at elevated temperatures. 22 Thermoplastic vulcanizates (TPVs), a unique type of TPE, possess a characteristic biphasic microstructure typically described as a “sea-island” morphology.23,24 In such systems, the particle size and crosslinking degree of the dispersed rubber phase play crucial roles in determining the stability of the sea-island structure and the overall mechanical performance of TPVs. Smaller rubber particles can markedly increase the interfacial area within the TPV system, thereby enhancing stress-transfer efficiency and enabling superior tensile properties, improved elastic recovery, and more stable viscoelastic responses. Furthermore, reducing the size of the rubber phase helps suppress microcrack initiation, leading to enhanced tear resistance and fatigue durability. However, excessive refinement of the rubber particles may significantly increase melt viscosity due to strengthened interfacial interactions, which can adversely affect processing flowability and potentially cause particle agglomeration, thereby diminishing the mechanical reinforcement typically achieved through particle size reduction.25–27
Shape memory polymers (SMPs) are a class of intelligent materials that can undergo reversible deformation in response to thermal, optical, electrical, magnetic, or chemical stimuli. They can be deformed and fixed into a temporary shape upon external stimulation and subsequently recover their permanent shape once the stimulus is removed.28–31 According to their activation mechanisms, SMPs can be categorized into thermally induced, photo-induced, electro-induced, magnetically induced, and chemically responsive types, among which thermally induced SMPs are the most extensively studied. 32 Thermally responsive SMPs (TSMPs) typically consist of a reversible phase and a switching phase: the reversible phase often features a chemically crosslinked network that provides the driving force for shape recovery, whereas the switching phase, governed by its melting temperature (T m ) or glass transition temperature (T g ), enables the fixation of the temporary shape.33–35 A typical dual-shape memory cycle includes four steps: deformation above the switching temperature (T sw ); cooling below T sw under load; unloading at low temperature while maintaining the temporary shape; and reheating to T sw to trigger shape recovery.36,37
Current studies on TPVs exhibiting shape memory behavior have focused mainly on conventional covalently crosslinked systems. Compared with irreversible covalent bonds, reversible noncovalent interactions play a critical role in enhancing the recyclability, self-healing capability, and shape-memory performance of thermoplastic elastomers. 38
A novel class of TSMPs based on a coordination-crosslinked EVA/NBR/LNBR TPV was prepared by dynamic vulcanization. The EVA/NBR/LNBR TPV exhibits a typical sea-island morphology. The incorporation of LNBR into the NBR phase of the EVA/NBR TPV markedly reduces the particle size of the dispersed NBR phase, significantly enhances the mechanical strength and elasticity of the TPV, and effectively improves its shape-recovery ratio (SR%). In addition, the effects of blend ratio, switching temperature, and strain amplitude on the shape memory behavior of the EVA/NBR/LNBR TPV were systematically investigated.
Experimental
Materials
Ethylene-vinyl acetate copolymer (EVA, grade 630, vinyl acetate content 15 wt%) was supplied by Tosoh Corporation, Japan. Nitrile butadiene rubber (NBR, grade 3305, acrylonitrile content 33 wt%) was obtained from PetroChina Lanzhou Petrochemical Company. Liquid nitrile butadiene rubber (LNBR, grade AZ8310, number-average molecular weight approximately 80,000, acrylonitrile content 30 wt%) was purchased from Dongguan Kaixuan Plastics Technology Co., Ltd., China. Anhydrous copper sulfate (CuSO4, analytical grade) was obtained from Tianjin Ruijiet Special Chemicals Co., Ltd. N-Isopropyl-N′-phenyl-p-phenylenediamine (antioxidant 4010NA) was supplied by Shen’ao Chemical Technology Co., Ltd.
Equipment and instruments
Instruments used in the experiments, along with their model numbers and manufacturers.
Preparation of EVA/NBR/LNBR TPVs
The compounding formulation of the NBR masterbatch (in parts per hundred rubber, phr) was as follows: NBR/LNBR 100 phr, CuSO4 10 phr, and 4010NA 1 phr.
Designation of TPVs with varying weight ratios.
Characterizations
Mechanical properties test
The mechanical properties of pure EVA, TPVs, and vulcanizates were measured using a servo-controlled tensile tester following the ASTM D412 standard.
Microscopy analysis
The surface morphologies of the etched EVA/NBR TPV and EVA/NBR/LNBR TPV samples were characterized by field-emission scanning electron microscopy (FE-SEM). The TPV samples were etched in a xylene solution at 100°C for 2.5 h selectively remove the EVA phase on the surface layer. The etched specimens were subsequently ultrasonically cleaned for 10 min and then vacuum-dried at 30°C until their mass remained unchanged.
For low-temperature brittle fracture observations, the TPV samples were immersed in liquid nitrogen until they were fully frozen, followed by manual fracturing. The resulting fracture surfaces were then examined. The tensile-fracture surfaces and longitudinal tensile surfaces of the TPVs were obtained from specimens broken during the mechanical tests. Before observation, a thin platinum layer was sputter-coated onto the sample surfaces to prevent charge accumulation.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was employed to investigate the melting and crystallization behaviors of EVA and EVA/NBR/LNBR TPVs to determine the shape-memory “switching” temperature. To minimize the effect of prior thermal history, the EVA/NBR/LNBR TPV samples were initially heated to 120°C and maintained at this temperature for 5 min. The samples were then cooled to 20°C at a rate of 20°C/min to record the crystallization curves. Thereafter, the samples were reheated to 120°C at a heating rate of 5°C/min to obtain the melting behavior curves.
Dynamic mechanical analysis
Dynamic mechanical analysis (DMA) measurements were conducted using a dynamic mechanical thermal analyzer. Temperature-sweep tests were performed in the range of −50°C to 120°C at a heating rate of 3°C/min under tensile mode. The testing frequency was 10 Hz with a strain amplitude of 0.5%.
Shape memory effect measurement
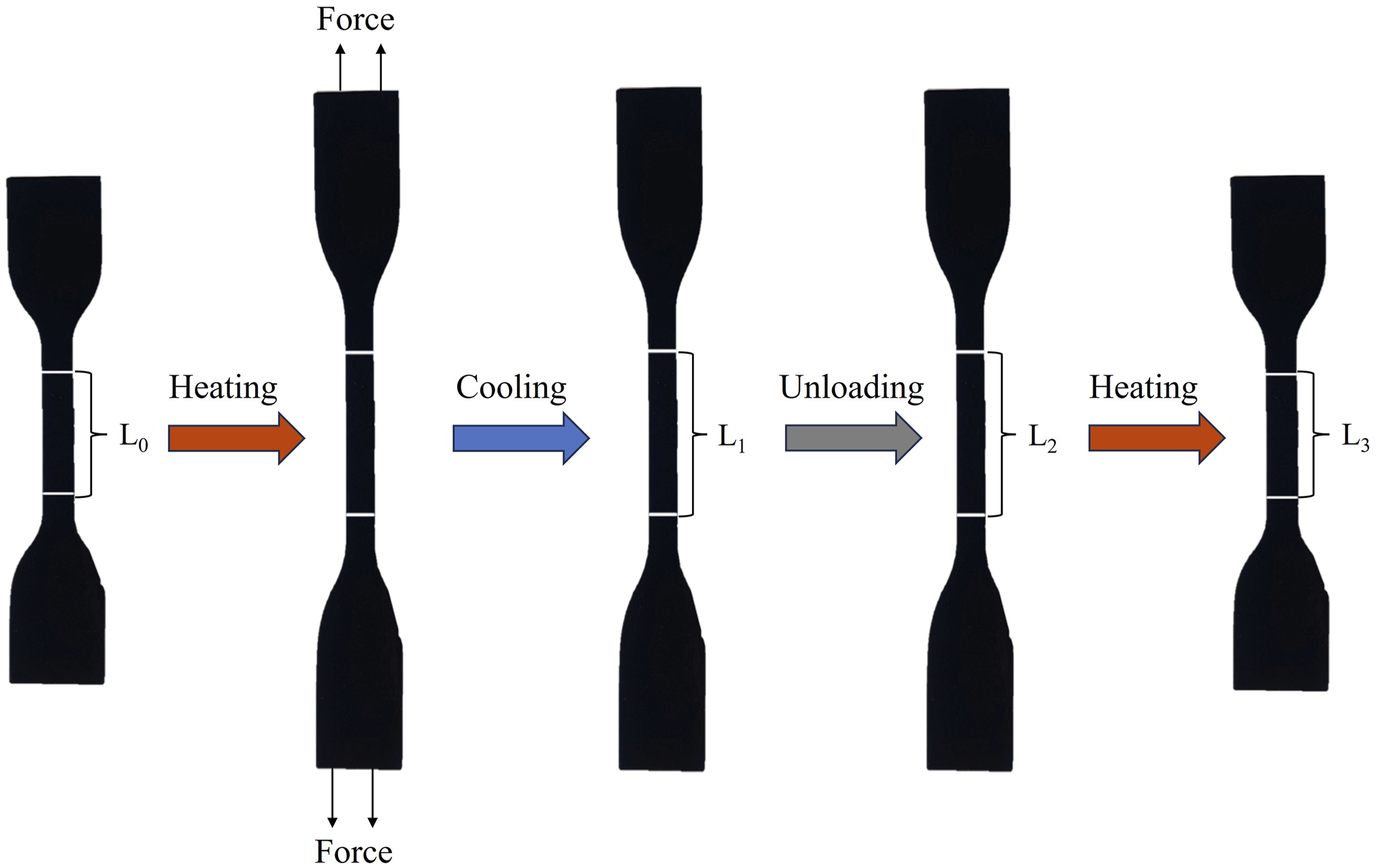
The shape memory performance was evaluated following the standardized procedure illustrated in Figure 1. (1) Two parallel lines with a spacing of approximately 20 mm were marked on the surface of a dumbbell-shaped EVA/NBR/LNBR TPV specimen, and the initial gauge length L
0
was measured and recorded. (2) The specimen was placed in a constant-temperature oven and heated to the preset deformation temperature (T
d
) for 10 min, after which it was stretched until the gauge length reached approximately 40 mm, a constant strain rate of 4 mm/s was applied during testing. (3) While maintaining the applied strain, the specimen was rapidly cooled in an ice-water mixture (0°C) for 5 min, and the corresponding gauge length L
1
was measured and recorded. (4) After releasing the load, the specimen was kept at ambient conditions (23 ± 2°C) for 24 h, followed by measurement and recording of the gauge length L
2
. (5) Finally, the specimen was placed in a constant-temperature oven and heated to the preset recovery temperature (T
r
) to trigger shape recovery, and the shape recovery time was uniformly set to 5 min, after which the final gauge length L3 was measured and recorded. To minimize experimental error, three specimens were tested at each pair of fixing and recovery temperatures, and the averaged values were used to ensure data reliability. Schematic portraying and illustration of SME measurement.
The shape memory performance was quantitatively evaluated using the shape-fixity ratio (SF%) and SR%, calculated according to equations (1) and (2):

Results and discussion
Mechanical properties of EVA/NBR/LNBR TPVs
For ease of description, abbreviations are used in this study to denote TPVs with different EVA/NBR/LNBR weight ratios. For example, E4N6 represents a TPV with an EVA-to-NBR weight ratio of 40/60; E40N54L6 denotes a TPV with EVA, NBR, and LNBR (number-average molecular weight of 80,000) weight ratios of 40/54/6, respectively.
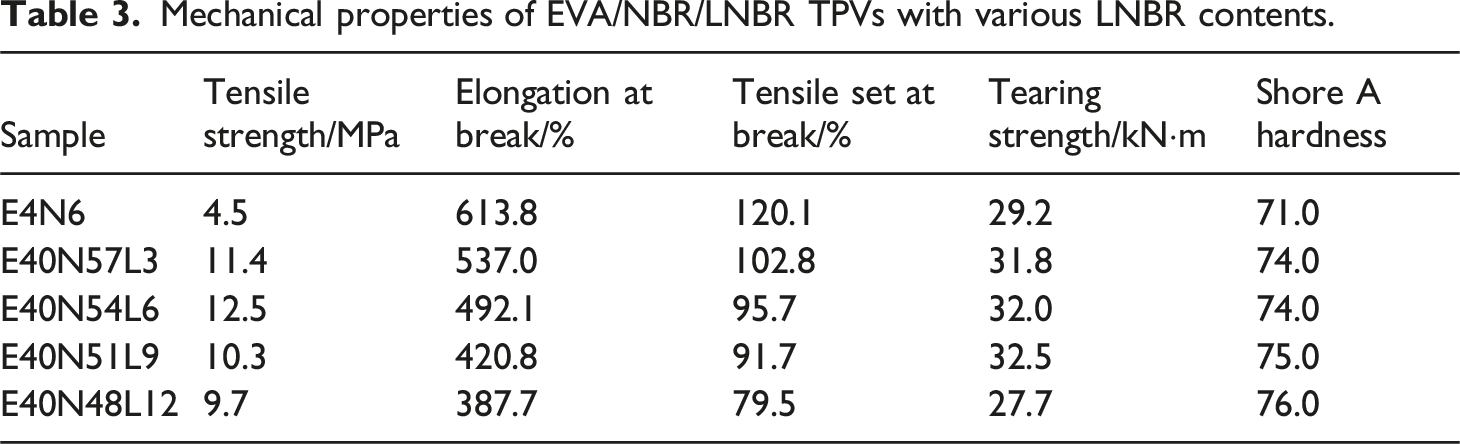
Mechanical properties of EVA/NBR/LNBR TPVs with various LNBR contents.
Mechanical properties of EVA, NBR/LNBR vulcanizates, and the series of EVA/NBR/LNBR TPVs.
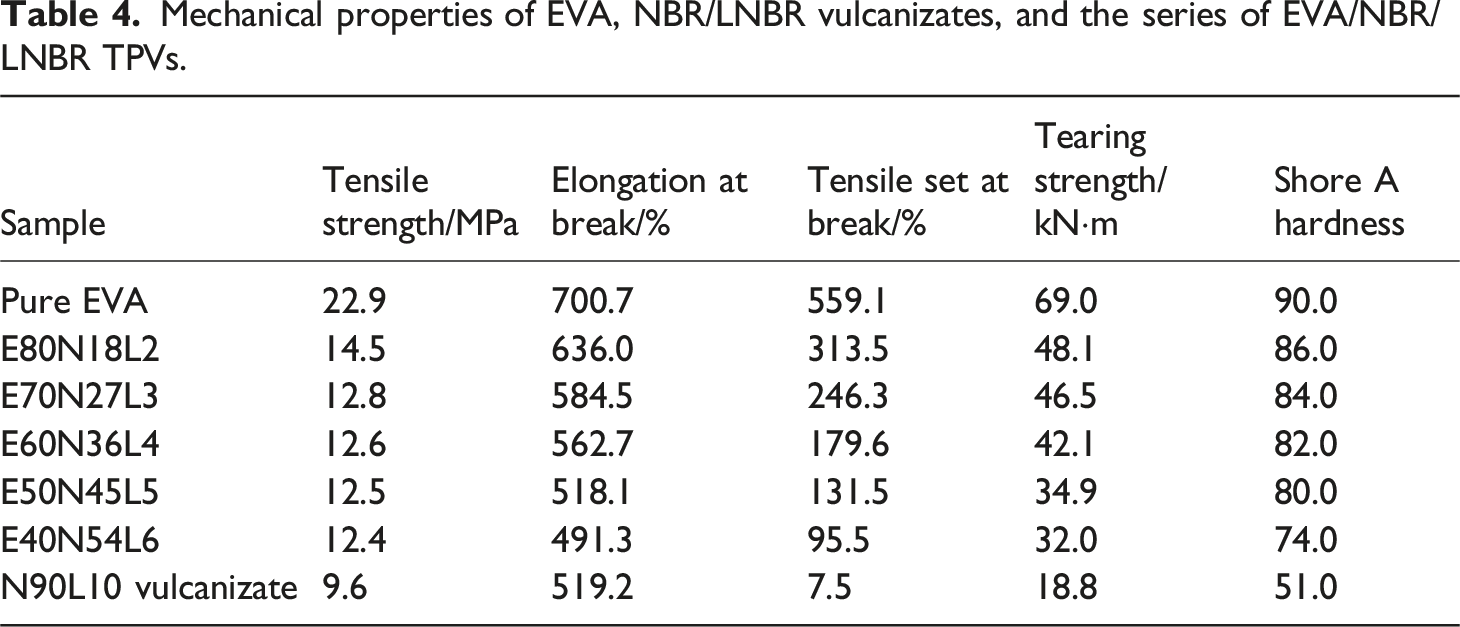
Figure 2(a) illustrates the stress-strain curves of the EVA/NBR/LNBR TPVs containing various amounts of LNBR. As shown in Figure 2(a), both the tensile strength and elongation at break exhibit clear trends with increasing LNBR content. With increasing LNBR loading, the elongation at break of the EVA/NBR/LNBR TPVs decreases continuously, while the tensile strength first increases and then decreases. The maximum tensile strength is achieved when the EVA/NBR/LNBR ratio is 40/54/6. Stress-strain curves of the EVA/NBR/LNBR TPVs series. (a) EVA/NBR/LNBR TPVs with various LNBR loadings; (b) EVA, NBR/LNBR static vulcanizates, and the EVA/NBR/LNBR TPVs series.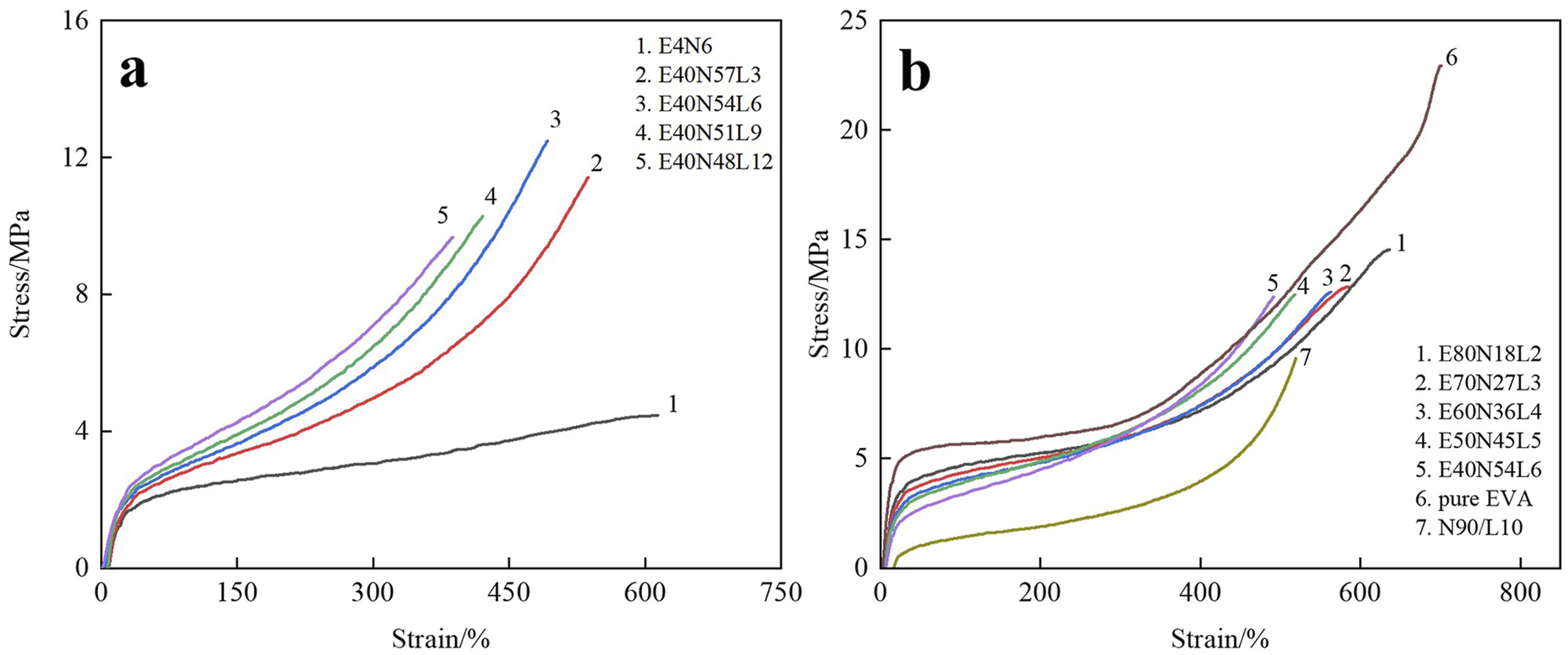
Figure 2(b) presents the stress-strain curves of EVA, NBR/LNBR static vulcanizates, and the series of EVA/NBR/LNBR TPVs. As shown in Figure 2(b), the variation trends of tensile strength and elongation at break for the EVA/NBR/LNBR TPVs can be clearly observed. With decreasing EVA content, the tensile strength of the EVA/NBR/LNBR TPVs shows a slight reduction. At the same time, the elongation at break exhibits a decreasing trend.
Microstructure of EVA/NBR/LNBR TPVs and EVA/NBR TPVs
Figure 3 presents the FE-SEM images of the E4N6 and E40N54L6 samples. Figure 3(a) and (c) show the surface microstructures of E4N6 and E40N54L6, respectively, after etching with xylene to remove the superficial EVA phase. As observed in Figure 3(a) and (c), both samples exhibit a typical “sea-island” biphasic morphology after etching. In the E4N6 sample, the exposed NBR vulcanized rubber particles possess diameters of approximately 7∼10 μm, whereas in the E40N54L6 sample, the diameters of the NBR/LNBR vulcanized rubber particles are markedly reduced to 3∼6 μm. This indicates that the incorporation of LNBR into the TPV system effectively decreases the particle size of the dispersed NBR rubber phase. FE-SEM images of E4N6 and E40N54L6. (a) Etched surface; (b) Tensile fracture surface; (c) Etched surface; (d) Tensile fracture surface; (e) Cryogenic brittle fracture surface; (f) Longitudinal tensile surface.
Figure 3(b) and (d) present the tensile fracture morphologies of the E4N6 and E40N54L6 samples, respectively. Comparative analysis shows that the fracture surface of the E4N6 sample exhibits pronounced ductile-fracture features. In contrast, the E40N54L6 sample displays a more uniform fracture morphology without any apparent signs of ductile tearing. Moreover, no significant detachment of NBR rubber particles is observed in the E40N54L6 sample, indicating that the interfacial adhesion between the NBR/LNBR rubber particles and the EVA matrix is substantially stronger than that in the E4N6 sample. This result further confirms that the incorporation of LNBR markedly enhances the interfacial compatibility between the EVA and NBR phases.
The morphology of the low-temperature brittle fracture surface of the E40N54L6 sample is shown in Figure 3(e). The fracture surface appears relatively smooth and flat, further confirming the strong interfacial interactions between the EVA matrix and the dispersed NBR/LNBR rubber phase. Figure 3(f) presents the longitudinal tensile surface morphology of the E40N54L6 sample, where an evident oriented structure formed within the EVA phase during tensile deformation can be clearly observed. The formation of this oriented structure is beneficial for stabilizing the deformed NBR/LNBR rubber particles, allowing the TPV to store substantial elastic energy and thereby providing the structural foundation necessary for the shape-memory recovery process.
DSC curves of EVA/NBR/LNBR TPVs
EVA is a semi-crystalline thermoplastic resin, and the melting temperature (T m ) of its crystalline regions is crucial for determining the deformation temperature (T d ) and the recovery temperature (T r ) in the dual-shape memory tests of EVA and EVA/NBR/LNBR TPVs. Therefore, investigating the melting behavior of EVA/NBR/LNBR TPVs through DSC is of significant importance.
Figure 4(a) shows the heating curves of the EVA/NBR/LNBR TPV series. A single melting peak is observed for both pure EVA and all EVA/NBR/LNBR TPVs, which is primarily attributed to the melting of the crystalline regions of EVA. Figure 4(b) presents the cooling curves of the EVA/NBR/LNBR TPV series. Distinct crystallization peaks are observed for all TPV samples, indicating that the EVA/NBR/LNBR TPVs possess good crystallization capability. On the one hand, the crosslinked NBR rubber phase acts as a nucleating agent and promotes the formation of EVA crystallites. On the other hand, the abundant NBR rubber particles impose substantial steric hindrance, which restricts the growth of EVA crystals. Thermal analysis curves of EVA and the series of EVA/NBR/LNBR TPVs. (a) Heating curves; (b) Cooling curves.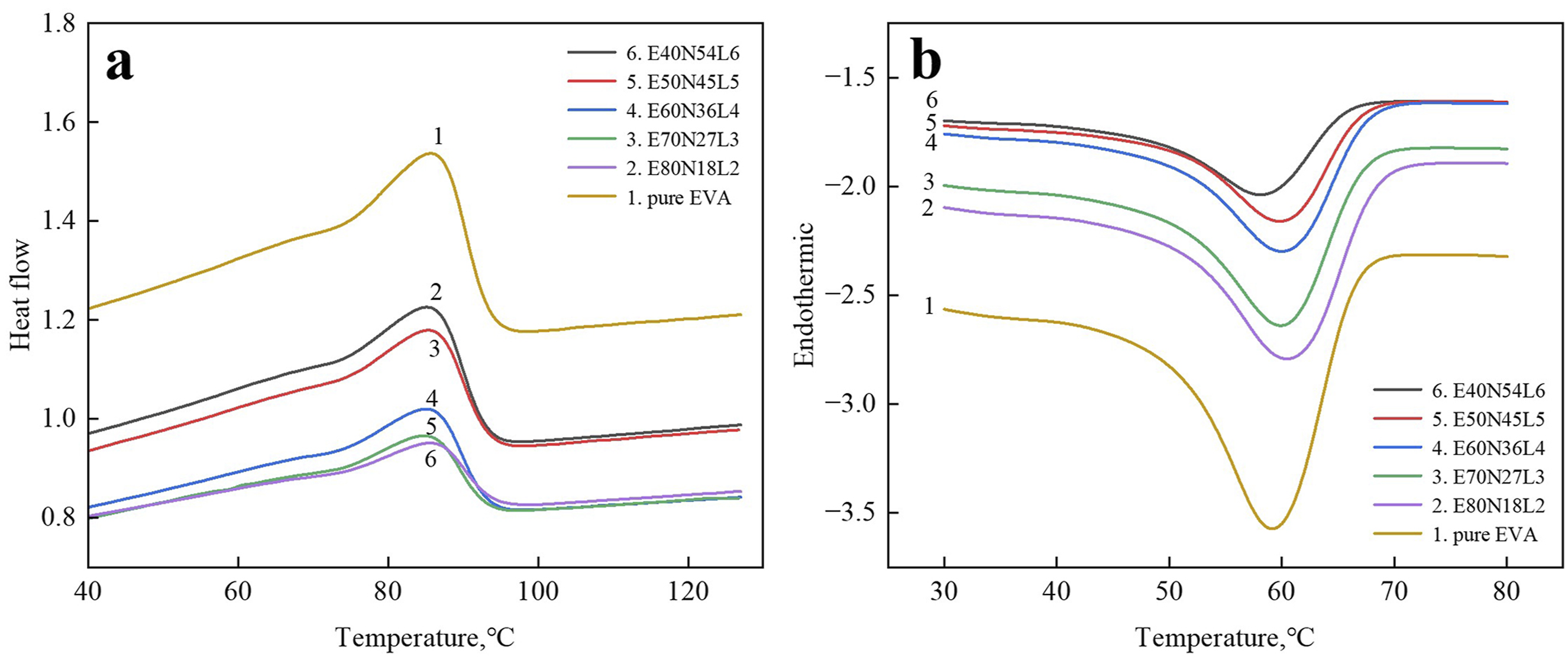
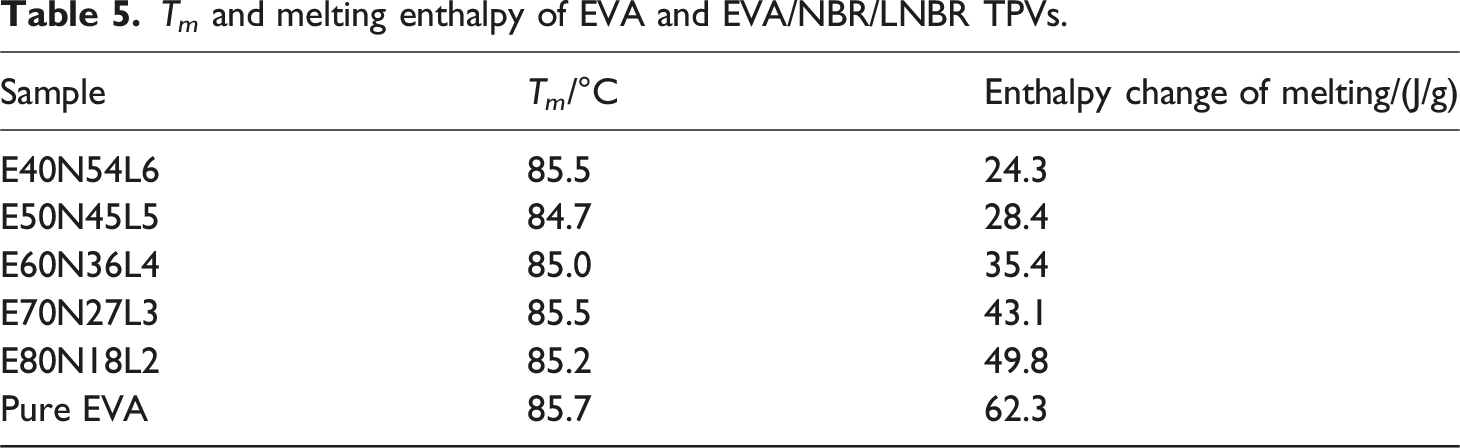
T m and melting enthalpy of EVA and EVA/NBR/LNBR TPVs.
DMA of EVA/NBR/LNBR TPVs
Figure 5(a) and (b) respectively show the relationships between temperature and the logarithm of the storage modulus (log E′) and loss modulus (log E″) for neat EVA, NBR/LNBR static vulcanizates, and EVA/NBR/LNBR TPVs. Figure 5(a) illustrates that the log E′ of all samples progressively declines as the temperature rises, reflecting a characteristic transformation from the glassy regime to the rubbery regime. Within the temperature interval of 0∼83°C, neat EVA shows the highest log E′ values, indicating its intrinsically high modulus. For EVA/NBR/LNBR TPVs, the log E′ increases with increasing EVA content, whereas increasing the proportion of NBR leads to a reduction in the rigidity of the TPVs and a noticeable improvement in their flexibility. When the temperature exceeds 83°C, the log E′ values of both EVA and the EVA/NBR/LNBR TPVs drop sharply, which is attributed to the melting of the crystalline domains of EVA at temperatures above approximately 83°C, resulting in a pronounced decrease in modulus. DMA curves of EVA, NBR/LNBR vulcanizate, and the series of EVA/NBR/LNBR TPVs. (a) Logarithm of storage modulus vs. temperature; (b) logarithm of loss modulus vs. temperature.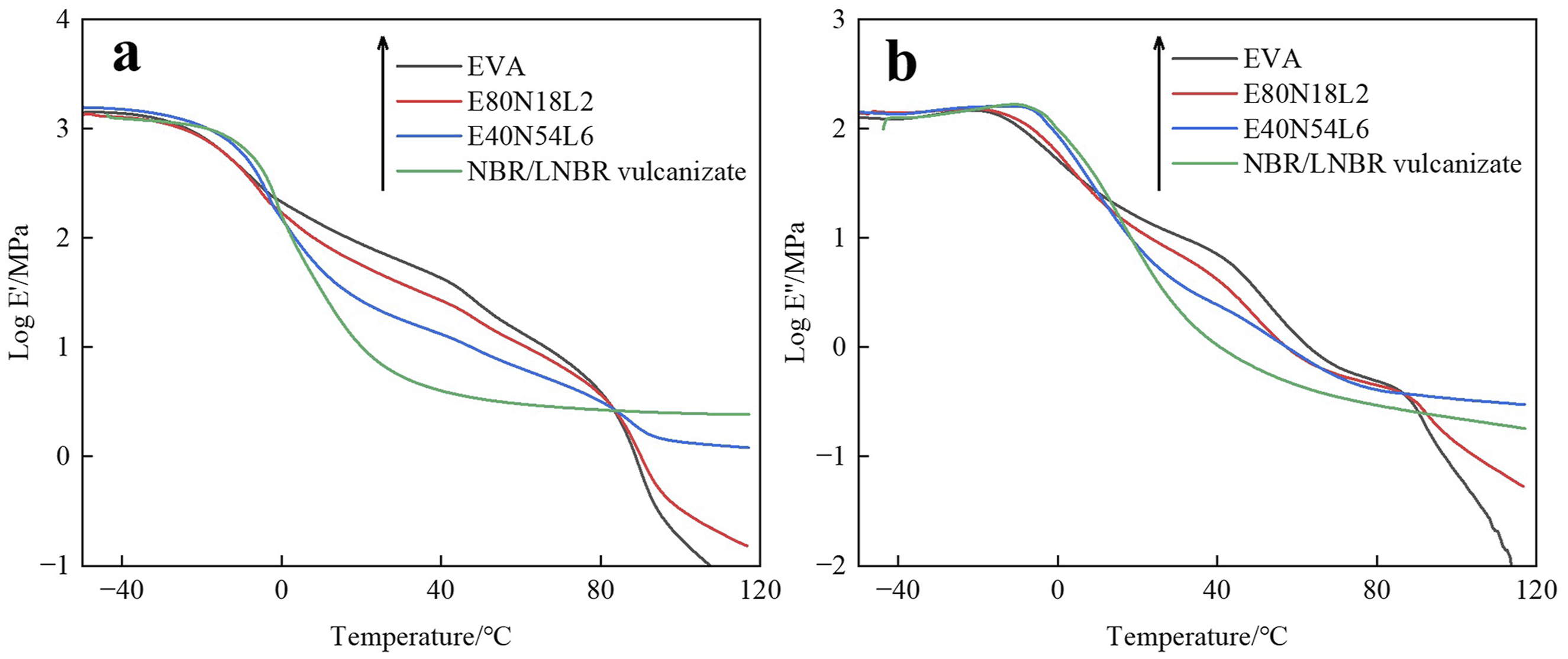
During the dynamic mechanical analysis test, when the temperature increases and approaches the glass transition temperature, the polymer chain segments begin to move. As a result, the internal friction of the material increases significantly, and the energy dissipation reaches a maximum, which is manifested as a pronounced peak in the Log Eʺ curve.
Figure 5(b) shows that pure EVA exhibits a peak at −21.9°C, while the NBR/LNBR vulcanizate presents a peak at −10.8°C. The peak temperatures in the loss modulus logarithm-temperature curves of the TPVs lie between those of EVA and the NBR/LNBR vulcanizate, and increase progressively as the EVA content decreases. The peak at −21.9°C can therefore be identified as the T g of EVA.
Figure 6 presents the temperature-dependent loss factor (tan δ) curves of neat EVA, NBR/LNBR vulcanizates, and the series of EVA/NBR/LNBR TPVs. As shown in Figure 6, for each material, the peak temperature of the tan δ curve is higher than that of the E″ curve, exhibiting the typical characteristics of viscoelastic materials. Moreover, as the EVA content decreases, the peak temperature of the tan δ curves shows a clear increasing trend. Temperature dependence of tan δ.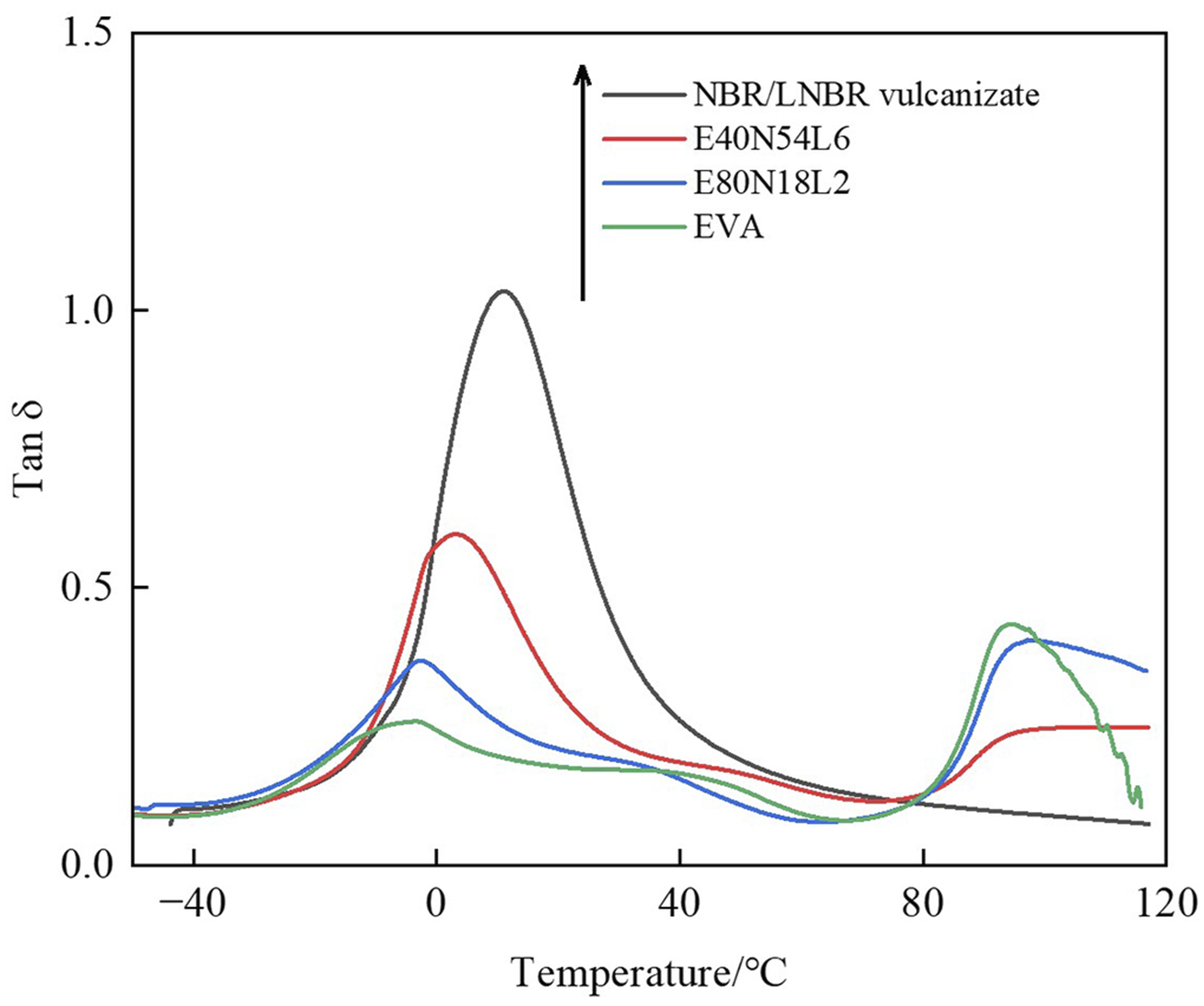
Shape memory property of EVA/NBR/LNBR TPV
Crystalline polymers are widely employed in the study of SMPs. Based on the DMA results in the section “DMA of EVA/NBR/LNBR TPVs”, EVA can be identified as possessing excellent shape-fixing capability. Therefore, EVA/NBR/LNBR TPVs constructed on a coordination-crosslinked network are expected to combine the shape-fixing ability of EVA with the shape-recovery capability of NBR, thereby enabling dual-shape memory behavior.
Figure 7 illustrates the shape-memory properties of EVA/NBR TPV, EVA/NBR/LNBR TPV, and NBR/LNBR vulcanizates at T
d
= 85°C and T
r
= 85°C. As shown in Figure 7, the shape-fixity ratio and shape-recovery ratio of E8N2 reach 95.4% and 95.5%, respectively, whereas those of E80N18L2 are 94.1% and 98.1%. These results indicate that introducing LNBR into the NBR phase slightly compromises the fixity ratio but significantly enhances the recovery ratio. Similarly, E4N6 and E40N54L6 exhibit the same variation trend, demonstrating that the incorporation of LNBR into NBR effectively modulates the shape-memory performance of TPVs. Combined with the microstructural observations in Figure 3, where the rubber-phase particle size is substantially reduced in EVA/NBR/LNBR TPVs, it can be inferred that the refined NBR dispersed phase provides a larger driving force for shape recovery, which is manifested macroscopically as an increased recovery ratio. Shape-memory performance of EVA/NBR TPVs, EVA/NBR/LNBR TPVs, and NBR/LNBR vulcanizates (Td = 85°C, Tr = 85°C). (a) Shape-fixity ratio; (b) Shape-recovery ratio.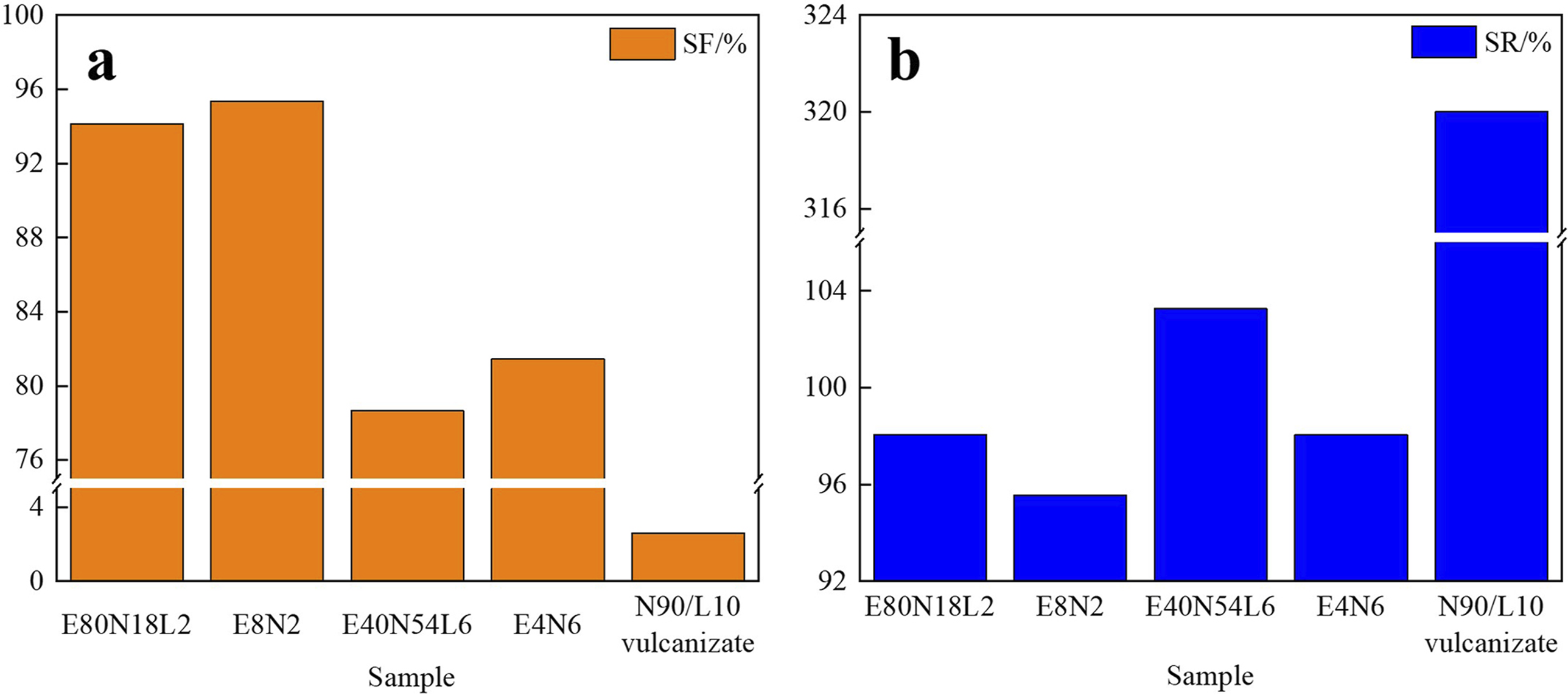
Temperature plays a crucial role in both the design and practical implementation of HSMPs. To gain deeper insight into the shape-memory behavior of EVA and EVA/NBR/LNBR TPVs, the shape-memory performance of neat EVA and a series of EVA/NBR/LNBR TPVs was systematically evaluated by varying T
d
and T
r
, thereby establishing the dependence of the SF% and SR% on T
d
and T
r
. Figure 8 presents the SF% and SR% of EVA/NBR/LNBR TPVs at different T
d
values with T
r
fixed at 85°C. As shown in Figure 8, under identical T
d
and T
r
conditions, the hardness of EVA is significantly higher than that of the NBR/LNBR vulcanizate. Therefore, increasing the EVA content raises the proportion of the continuous EVA phase, thereby enhancing the overall hardness and rigidity of the TPV. The increased EVA content also improves the ability of EVA crystallites to lock the dispersed NBR/LNBR rubber particles, enabling the material to better maintain the temporary shape after deformation and thus increasing the shape-fixation ratio. Meanwhile, the relative proportion of the rubber phase decreases, reducing the storage and release of elastic potential energy, which results in a lower shape-recovery ratio. SF% and SR% of EVA/NBR/LNBR TPVs at different Td (Tr = 85°C)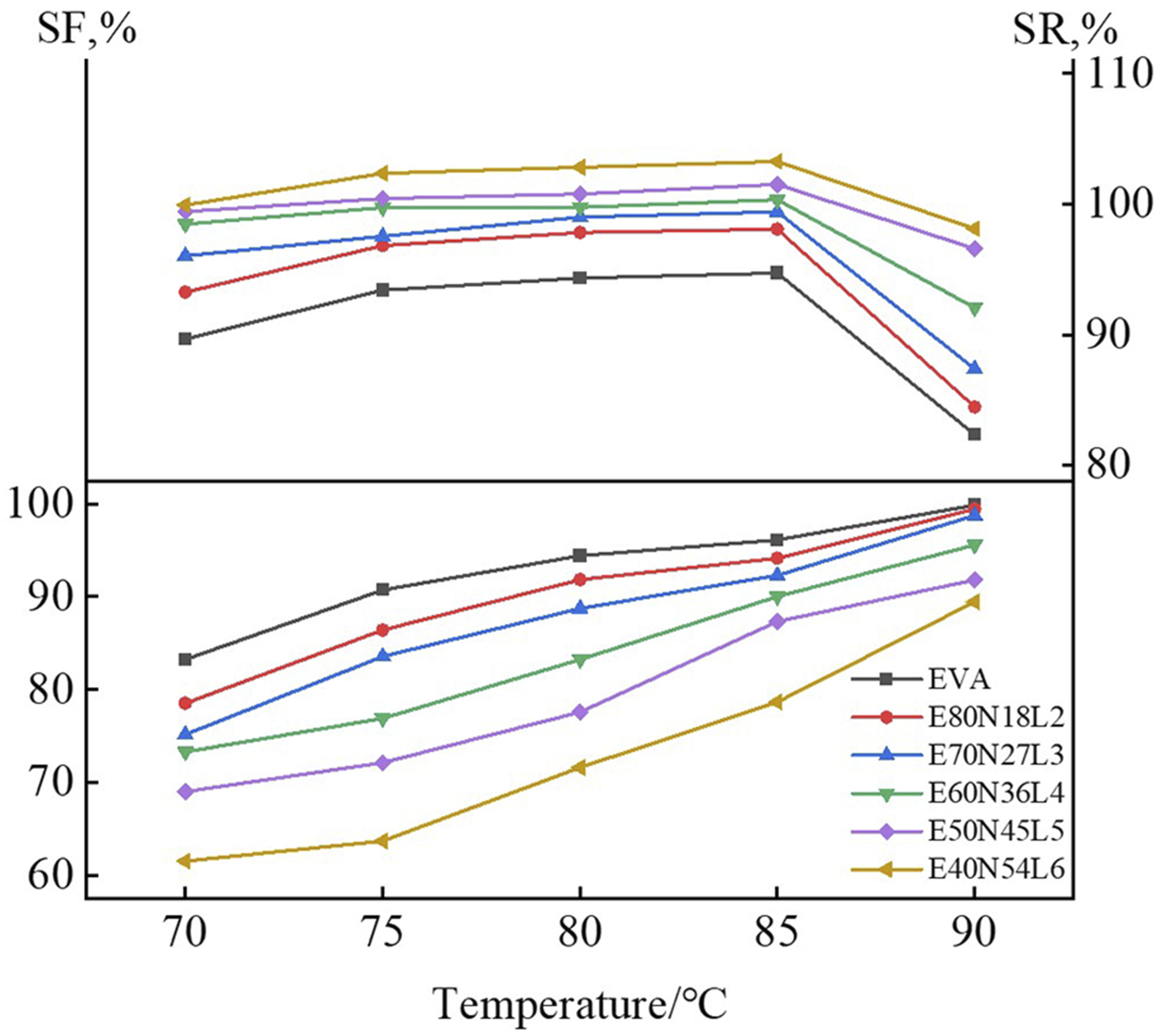
The setting of T d exerts a significant influence on both SF% and SR%. With increasing T d , the SF% of EVA and EVA/NBR/LNBR TPVs gradually increases; for instance, the SF% of E80N18L2 reaches 94.1% at 85°C. Meanwhile, the SR% of EVA and EVA/NBR/LNBR TPVs also increases and attains a maximum at 85°C. When the temperature exceeds the melting point of EVA (i.e., 90°C), the SR% of EVA/NBR/LNBR TPVs decreases, as most EVA crystals are in the molten state, reducing the strength of the EVA phase. Consequently, insufficient stress can be transferred to deform and fix the NBR rubber particles. The slightly deformed NBR particles cannot provide adequate recovery force for the deformed EVA/NBR/LNBR TPV samples, thereby weakening their shape-recovery capability. Notably, the SR% of E80N18L2 reaches 98.1% at 85°C, indicating that this TPV exhibits not only excellent shape-fixation ability but also outstanding shape-recovery performance.
As T d increases, the chain-segment mobility of EVA macromolecules is enhanced, which facilitates deformation and orientation of the EVA phase. Meanwhile, the oriented EVA structure can more effectively transfer stress to the NBR/LNBR rubber particles, enabling their deformation to be maximally fixed, which is beneficial for subsequent shape recovery. However, when T d exceeds the T m of EVA, most EVA crystals melt, leading to a loss of phase strength and rendering the EVA phase incapable of imposing sufficient stress on the NBR/LNBR particles to deform and fix them. As a result, the slightly deformed NBR/LNBR particles fail to provide adequate recovery force for the EVA/NBR/LNBR TPV samples, thereby diminishing their shape-recovery capability.
Moreover, at a fixed T d , the SF% of EVA/NBR/LNBR TPVs increases with increasing EVA content, yet it remains inferior to that of neat EVA. In contrast, the SR% of EVA/NBR/LNBR TPVs rises with increasing NBR content and is markedly higher than that observed for neat EVA. These results indicate that the EVA phase plays the dominant role in shape fixation in EVA/NBR/LNBR TPVs, whereas the NBR/LNBR phase makes a substantial contribution to the shape-recovery process.
Figure 9 shows the shape recovery ratios of the EVA/NBR/LNBR TPVs at various T
r
when the T
d
is fixed at 85°C. As illustrated in Figure 9, the SR% values of neat EVA and the EVA/NBR/LNBR TPV series increase progressively with increasing T
r
. With the rise in recovery temperature, the strength of the EVA phase decreases, thereby weakening its constraint on the NBR/LNBR rubber particles. This facilitates the release of the elastic potential energy stored in the deformed NBR/LNBR rubber particles, consequently enhancing the shape recovery capability of the EVA/NBR/LNBR TPVs. When T
r
reaches 85°C, the SR% of E80N18L2 attains 98.1%, which is significantly higher than that of neat EVA, demonstrating its excellent shape recovery performance. SR% of EVA/NBR/LNBR TPVs at different Tr values (Td = 85°C).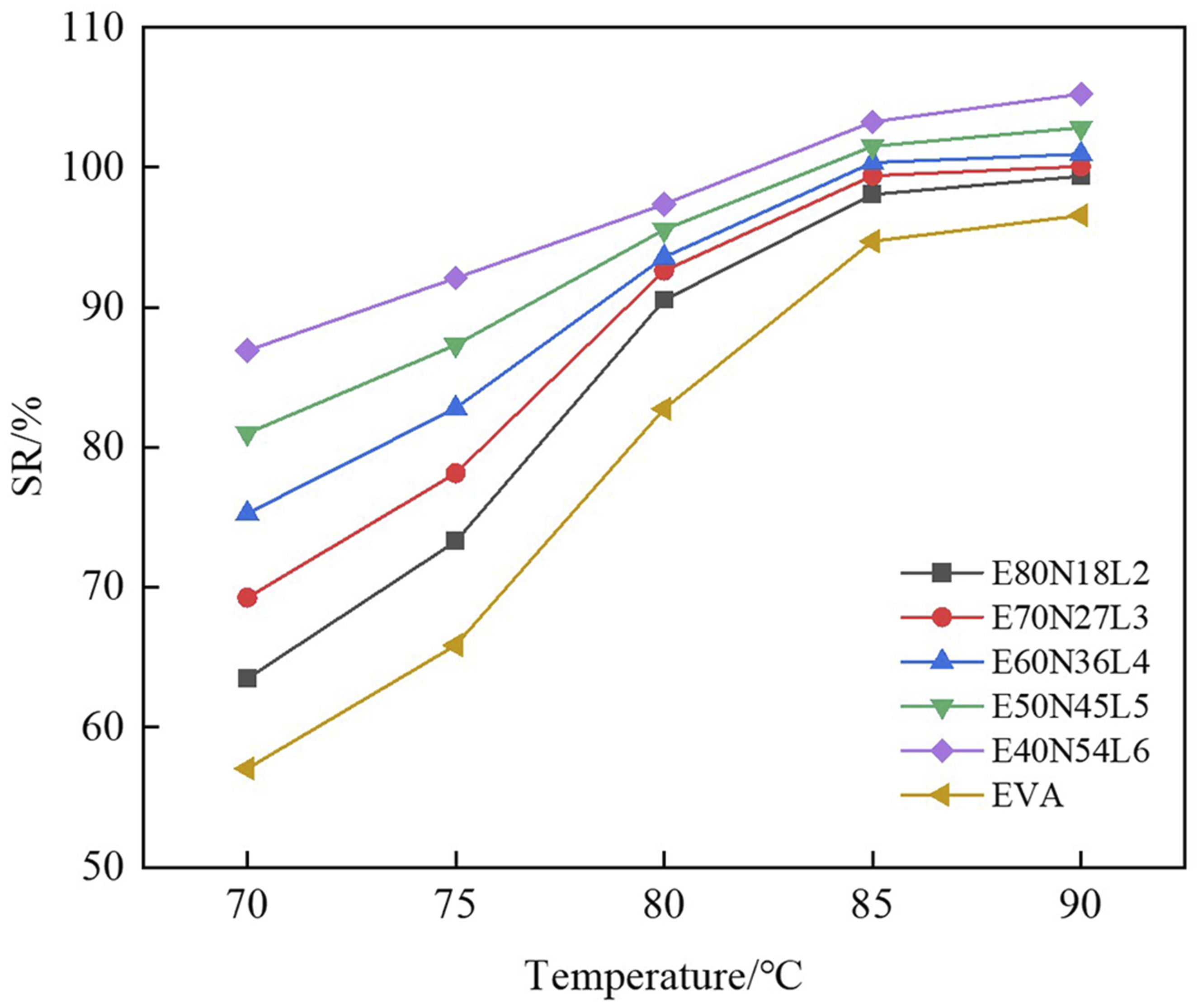
Figure 10 presents the SF% and SR% of E80N18L2 under different applied strains, with both the T
d
and T
r
fixed at 85°C. As shown in Figure 10, both SF% and SR% decrease progressively with increasing strain amplitude, indicating that the shape-memory performance of EVA/NBR/LNBR TPVs is closely related to the complexity of the imposed deformation. Larger deformations not only require stronger shape-fixing capability but also tend to induce greater permanent deformation, thereby leading to a continuous reduction in both SF% and SR%. Nevertheless, within the studied system, SF% remains above 91.7%, while SR% still exceeds 95.9%. SF% and SR% of E80N18L2 under different strains (Td = 85°C, Tr = 85°C).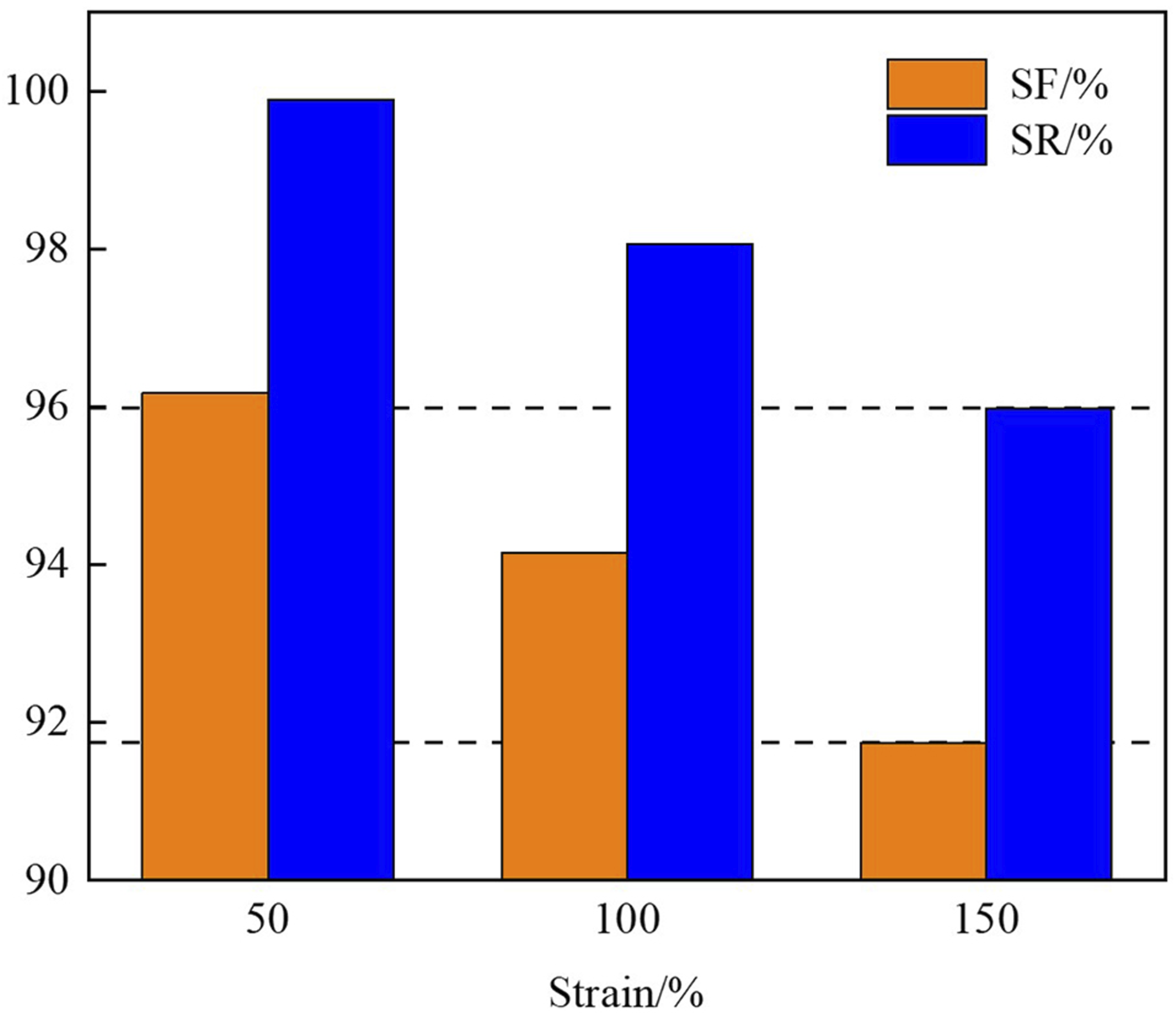
To provide a more intuitive visualization of the shape-recovery process of the EVA/NBR/LNBR TPV, three distinct temporary shapes were additionally designed. Figure 11 illustrates the shape-recovery behavior of E80N18L2 at T
d
and T
r
both set to 85°C, with recovery conducted in silicone oil maintained at the same temperature. As shown in Figure 11, specimens of E80N18L2, fixed into different temporary shapes, were able to fully return to their original configuration within 20 seconds. However, slight differences in recovery time were observed among the shapes. This indicates that the complexity of the temporary shape influences the recovery kinetics of E80N18L2. It is noteworthy that the EVA/NBR/LNBR TPV exhibits multiple programmable temporary shapes as well as a desirable medium-speed response, suggesting its potential applicability in fields such as temperature-responsive sensors.
39
Shape recovery behavior of E80N18L2 at Td = 85°C and Tr = 85°C.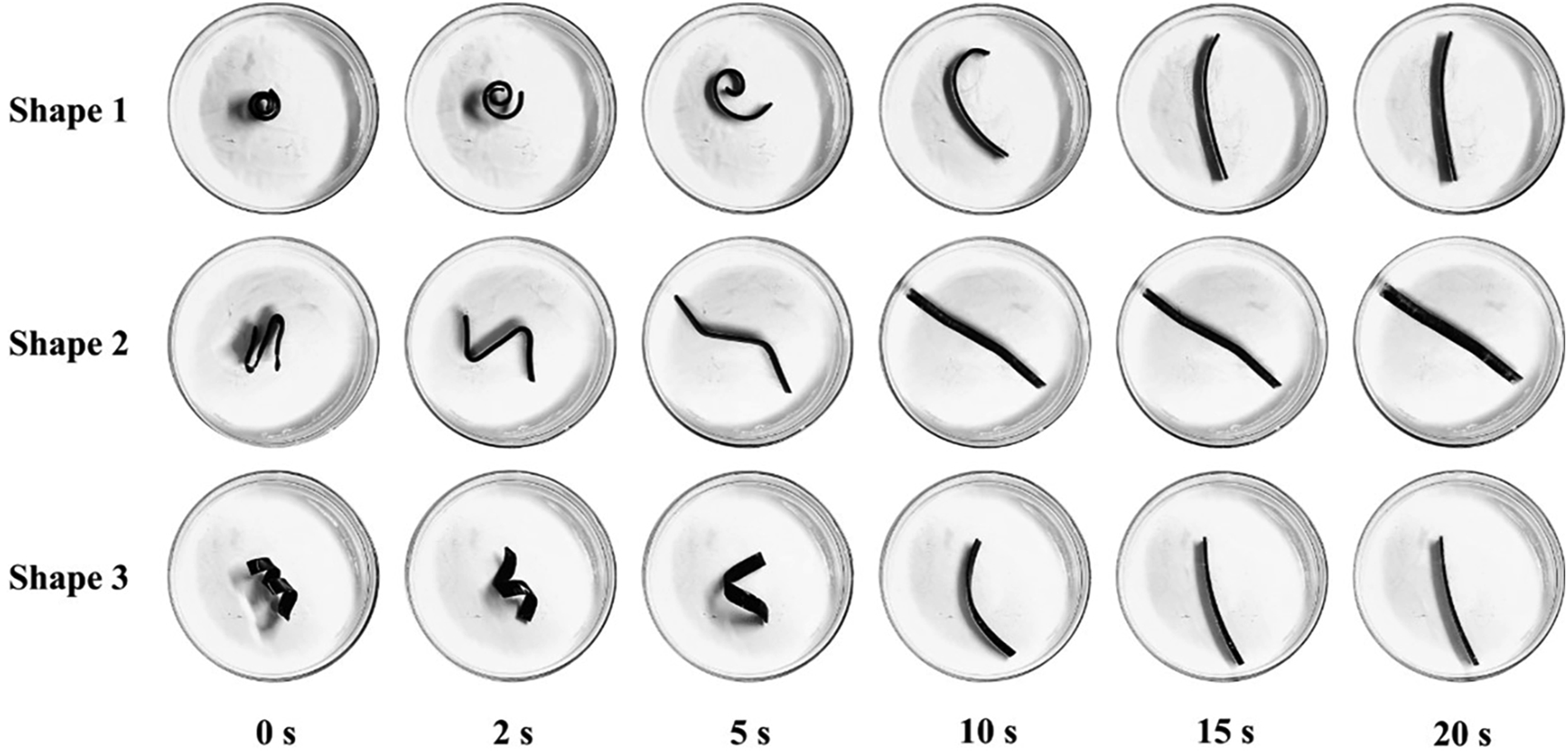
Based on the above results, a dual-shape memory mechanism is proposed for the EVA/NBR/LNBR TPV system. As illustrated in Figure 12, the schematic diagram reveals the microstructural evolution of the material during the shape memory process. According to the findings presented in Section 3.6, the EVA/NBR/LNBR TPV exhibits a typical “sea-island” biphasic morphology. When the T
d
approaches the T
m
of the EVA thermoplastic matrix, the rigidity of the TPV gradually decreases, allowing the material to transition from its original shape to a temporary configuration. During this deformation process, the NBR/LNBR rubber particles dispersed within the EVA matrix undergo forced deformation under the applied external stress. Subsequently, when the deformed EVA/NBR/LNBR TPV is cooled to a temperature far below the T
m
of the EVA phase, crystallization of the continuous EVA matrix occurs, and the deformed NBR/LNBR rubber particles become immobilized within the rigid EVA framework. This process stores the elastic energy required for shape recovery. Upon reheating to a temperature near the T
m
of the EVA matrix, the oriented crystalline domains of EVA relax. At the same time, the confined NBR rubber particles release the stored elastic potential energy. These two effects act in concert to drive the material back to its original configuration from its temporary shape. In this study, the shape-memory performance was evaluated based on a single-cycle test, which has certain limitations and cannot fully reflect the long-term cyclic stability of the material. However, the applied programming strain is relatively small and has limited influence on the internal TPV structure. Therefore, the material is expected to maintain good structural stability during cyclic deformation, allowing the shape-memory performance to be preserved to a certain extent. Schematic illustration of the dual-shape memory mechanism of the EVA/NBR/LNBR TPV.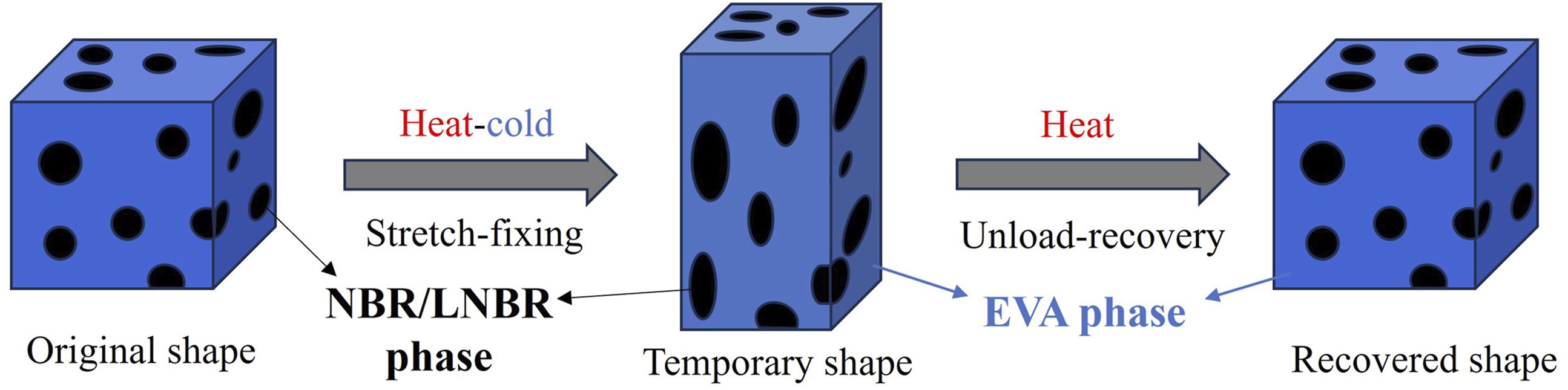
Conclusions
Introducing LNBR into the NBR phase of the coordination-crosslinked EVA/NBR TPV markedly enhances the interfacial compatibility between EVA and NBR, significantly reduces the rubber-phase particle size within the TPV, and substantially improves its mechanical strength. When the EVA content is fixed, and the mass ratio of NBR to LNBR is varied, the tensile strength of the EVA/NBR/LNBR TPV exhibits the most pronounced increase at an NBR/LNBR ratio of 90/10. When the NBR/LNBR mass ratio is fixed, increasing the EVA content leads to simultaneous increases in tensile strength, tear strength, hardness, elongation at break, and permanent deformation at break of the EVA/NBR/LNBR TPV. Investigations of the shape-memory properties reveal that the introduction of LNBR into NBR slightly compromises the shape-fixity ratio, while significantly enhancing the shape-recovery ratio of the TPV. This demonstrates that LNBR effectively tunes the shape-memory performance of the EVA/NBR/LNBR TPV system. When both T d and T r are set at 85°C, the shape-fixity ratio and shape-recovery ratio of E80N18L2 reach 94.1% and 98.1%, respectively. Furthermore, E80N18L2 specimens programmed into different temporary shapes can fully recover to their original shape within 20 s, indicating that the coordination-crosslinked EVA/NBR/LNBR TPV exhibits excellent shape-memory capability.
Footnotes
Author’s note
This manuscript has not been published elsewhere and it has not been submitted simultaneously for publication elsewhere.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by the Shandong Provincial Natural Science Foundation, China (ZR2021ME028).