
Abstract
Phenolphthalein polyethersulfone (PES-C)/silica (SiO2) composite nanofibrous membranes were prepared via solution blowing. The spinning solutions were prepared by mixing a solution of PES-C in dimethylacetamide with different amounts of colloidal SiO2 in ethylene glycol. Attenuated total reflectance–Fourier transform infrared spectroscopy, wide-angle X-ray diffraction, scanning electron microscopy, thermal gravimetric analysis, and water contact angles were conducted to characterize the properties of composite nanofibrous membranes. The results showed that the thermal properties and surface wettability were improved by the addition of appropriate amount of nano-SiO2. Furthermore, permeation fluxes of pure water and the filtration of starch suspension were measured to evaluate the antifouling property of the PES-C/SiO2 composite membranes.
Introduction
Phenolphthalein polyethersulfone (PES-C) as an engineering plastic, which contains cyclic side groups in the backbone, has been widely used as membrane materials such as gas permeation, 1 ultrafiltration membrane, 2 fuel cells, 3 and other composite membranes preparation. 4,5 It possesses good solubility, high glass transition temperature, excellent thermal stability, and mechanical properties, but the poor hydrophilicity of these membranes often leads to serious membranes fouling problem that would limit their application in the filtration field. There have been many reports about improving the hydrophobicity of the membranes, 6 –8 among these, adding nanoparticles into the membranes that were prepared via phase inversion process is one of the proverbial ways. 9 –15
Most work about the preparation of polymer/nanoparticles composite nanofibers focused on the enhancement of the thermal properties and mechanical properties. 16 –19 In addition, there have been research about blending nanoparticles to the fibers to fabricate superhydrophobic composite membranes due to the surface roughness of the membranes were reinforced or the nanoparticles were modified into hydrophobic. 20 –23 But we believed that the right amount of hydrophilic nanoparticles that were well dispersed on the surface of fibers would enhance the surface wettability of the mats. 24
There are only few reports about blending the hydrophilic nanoparticles into the fibers to improve the surface wettability of the membranes. Nano-silica (SiO2) have been utilized to enhance the hydrophilicity of membranes due to the numerous hydroxyl groups on the surface, but they are easy to agglomerate together because of their high surface energy and instability. In this article, we prepared PES-C/SiO2 composite nanofibrous membranes via solution blowing (SB) technology; two kinds of colloidal SiO2 that were dispersed in ethylene glycol (EG) and water, respectively, as the source of SiO2 were selected to mix into the spinning solution. Then, the nanofibrous membranes were characterized by Fourier transform infrared (FTIR) spectroscopy, wide-angle X-ray diffraction (WAXD), scanning electron microscopy (SEM), thermal gravimetric analysis (TGA), water contact angle (WCA), and so on. Finally, the permeation flux of pure water and the filtration of starch suspension were measured to evaluate the antifouling property of the composite membranes.
Experiment
Materials
Bis(4-chlorophenyl) sulfone (99%) was purchased from Alfa Aesar Chemical Co. Ltd, China.
Phenolphthalein (99%), potassium carbonate (99%), and starch soluble (analytical grade purity) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Sulfolane (99%) was supplied by Sigma-Aldrich Trading Co. Ltd, Shanghai and was purified by active carbon and stored over 0.4 nm molecular sieve.
N,N-Dimethylacetamide (DMAc, 99%) and toluene (99.5%) were acquired from Ling Feng Chemicals Co., Ltd (Shanghai, China) and was purified by distillation and dried by 0.4 nm molecular sieve.
Colloidal SiO2 (30% by weight in EG, 20 nm) was from Alfa Aesar and colloidal SiO2 (30% by weight in water, 30 nm) was from Qingdao Fuso Refining & Processing Co., Ltd; they were used as received without further purification.
All other chemicals were in an analytical grade and used as received.
Preparation of spinning solution
PES-C was prepared in the laboratory 25 and the inherent viscosity (η) was about 0.65 dL/g. Certain amount of colloidal SiO2 in EG was firstly added into DMAc and thereafter ultrasonicated for 30 min to ensure complete dispersion of the nanoparticles. Then, the identical weight of PES-C was mixed and dissolved in DMAc under mechanical stirring for 10 h to form a uniform homogeneous spinning solution at room temperature, after that they were under vacuum at 25°C for 24 h to be degassed. The colloidal SiO2 in water was blended with the PES-C spinning solution following the same processing as above. The compositions of PES-C/SiO2 spinning solutions are listed in Table 1.
The composition of membrane spinning solutions.
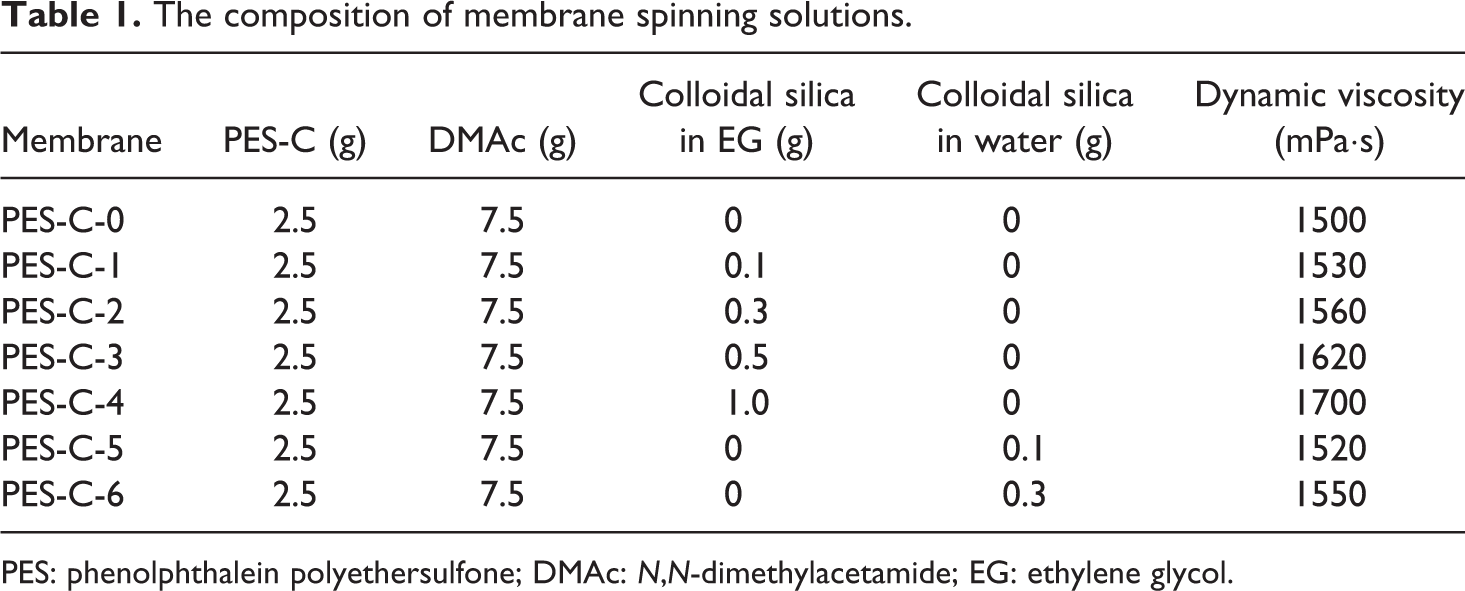
PES: phenolphthalein polyethersulfone; DMAc: N,N-dimethylacetamide; EG: ethylene glycol.
Solution blown spinning
The PES-C nanofibrous membranes were fabricated via homemade SB apparatus. The spinning solution was squeezed out through a 0.5 mm inner diameter needle at a speed of 3 ml/h under 0.1 MPa nitrogen pressure. The nanofibers were deposited on the rotating collector to form a fibrous mat, and the collecting distance was 20 cm. Vacuum drying was adjusted at 80°C for 12 h to remove the residual solvent. The thickness of PES-C/SiO2 composite membranes was about 250 µm, and the apparent density of the membranes was about 27–28 g/m2. The SB apparatus is shown in Figure 1.
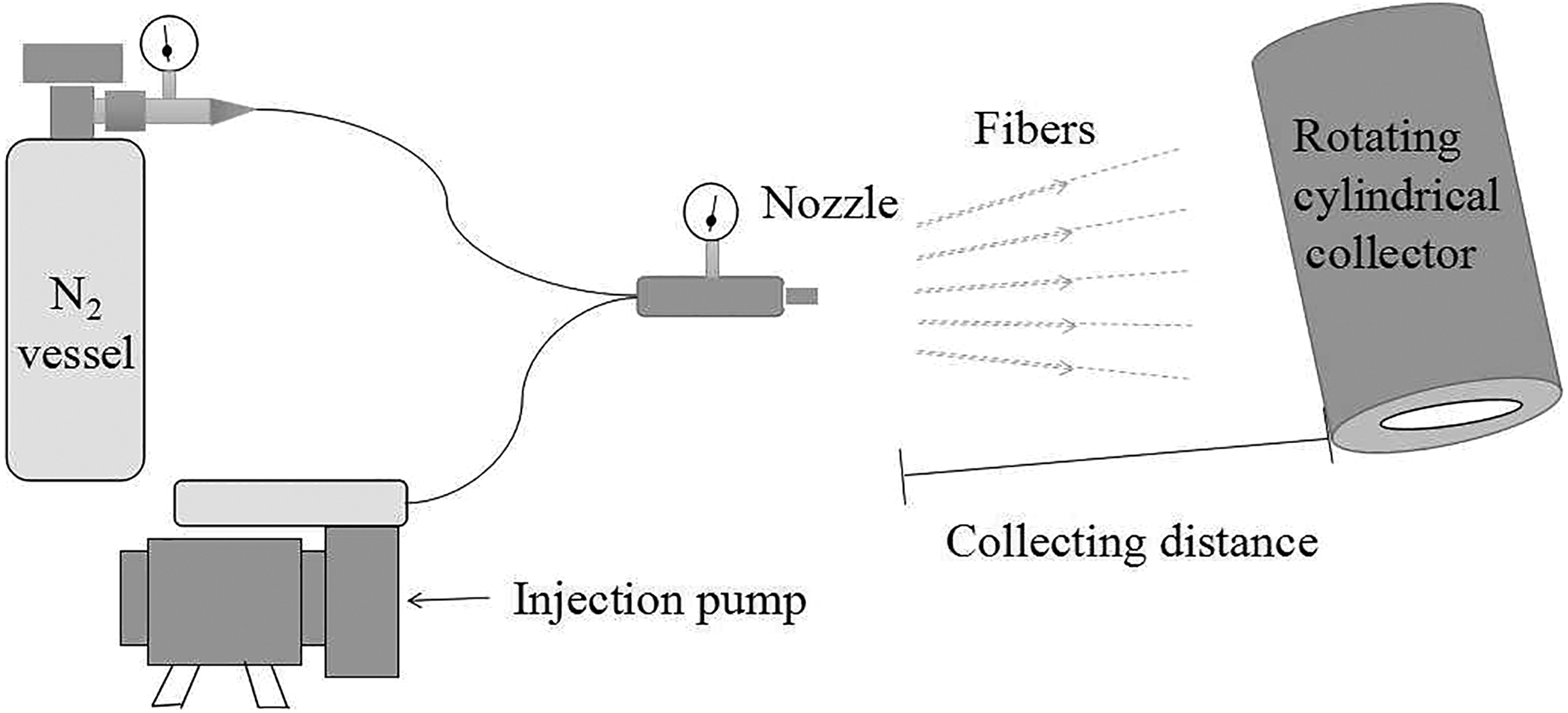
Schematic representation of the SB setup. SB: solution blowing.
Filtration experiment
Dead-end filtration was performed by homemade membrane permeation test apparatus that consisted of an air pump, gas flow meter, recipient container, and an effective area (membrane) of 7 cm2. Before the filtration tests, the membranes were immersed in deionized water for 15 min to be wetted. Then, the flux tests were operated at 5 kPa, and the initial water flux (F) was examined as Jw1. The pure water flux was evaluated using the following equation

where Jw1 is the pure water flux of the membrane (L/m2·h), Δt is the permeation time (h), A is the effective membrane area (m2), and V is the volume of permeation water during the permeation time (L). At least, three samples of each membrane were selected to measure the water flux, and the mean value was reported.
Antifouling properties tests
After the pure water flux (Jw1) was tested first, the starch suspension with the concentration of 2 g/L was chosen as foulant particles to evaluate the antifouling properties of the membranes. After 60 min of starch suspension filtration under ambient pressure, the membranes were simply backwashed with tap water for about 5 min, and then the pure water flux was measured again which was examined as Jw2 at 5 kPa. At least, three samples of each membrane were measured, and the mean value was obtained to minimize the test errors. The fouling-resistant ability of the membranes was evaluated by flux recovery ratio (FRR) using the following equation
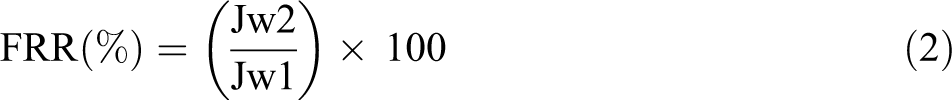
Characterization
Inherent viscosity (η) was measured in DMAc at a concentration of 0.5 g/dL at 25°C using a Ubbelohde viscosimeter (Shanghai Shuo Optical Electronics Technology Co. Ltd).
The average thickness of the nanofibrous membranes was measured using a digital micrometer (Shanghai Precision Instrument Co. Ltd).
The morphology of the nanofibrous membranes was examined by SEM (Quanta 250, Czech Republic FEI Co.). The mean diameter of the nanofibers was obtained by measuring the diameter of 50 random fibers chosen from their SEM images using the Image J, version V1.48u, software.
FTIR spectra were recorded on a Nicolet Nexus 8700 spectroscope (Thermo Fisher Scientific Co., USA) with the range of 4000–400cm−1 to characterize the chemical structure and characterization of the membranes.
WAXD was performed on a D8 Advance X-ray diffractometer (Bruker, Germany) with 2θ ranging between 5° and 60°.
TGA was conducted with a Netzsch TG209F1 Iris instrument (Netzsch Scientific Instruments Trading (Shanghai) Co., Ltd, Germany) carrying out on approximately 5 mg at a heating rate of 10 K/min from 50°C to 800°C under nitrogen atmosphere at a purge rate of 20 mL/min.
WCA measurements were performed using a contact angle goniometer Kino SL200B (USA Kino Industry Co., Ltd, USA) by the sessile drop method at room temperature. The image was recorded immediately (about 3 s), and the mean WCA results were obtained by measuring at least 10 spots of each membrane to get a reliable value.
The original and composite nanofibrous membranes were cut into 2 × 2 cm2, then immersed in n-butanol for 3 h, and finally the residual solvent on the membranes surface is wiped using a filter paper. The porosity of each substrate (P) was calculated using the following equation

where ρ is the density of n-butanol (0.81 g/cm3), Vm is the volume of membranes, and m0 and mw is the weight of membranes before and after immersion.
The size of the starch soluble was measured by the Malvern Mastersizer Particle size analyzer (MS2000, Malvern Instruments Ltd, UK).
The original and composite nanofibrous membranes were cut into 3 × 3 cm2 for porometry measurement and a CFP-1100-AI capillary flow porometer (Porous Materials Int. Co., USA) was used to measure the pore size and pore size distribution of the membranes, and the test was repeated three times to obtain an average value.
Results and discussion
Morphology of membranes
SEM images indicating the surface morphology and corresponding fiber diameters of the original and composite membranes are shown in Figure 2. The compositions of spinning solutions and corresponding viscosity are shown in Table 1. At high magnification (10,000×), it could be seen that the nano-SiO2 dispersed well on the surface of the fibers, and the morphology of initial PES-C nanofibers and PES-C/SiO2 (colloidal SiO2 in EG) composite nanofibers were similarly smooth and defect-free as shown in Figure 2(a) to (e). The fiber diameter increased with the raising content of the nano-SiO2 as shown in Figure 2, which may be due to the increase of solution viscosity. The result showed that good dispersion of SiO2 nanoparticles was in contrast to that in the case of other SiO2 composite fibers published in the literature, where nano-SiO2 particles agglomerated heavily. 16,17 The average diameter of the original PES-C fibers was about 0.44 µm, and it gradually increased to about 0.50, 0.53, 0.60 and 0.65 µm, respectively, ranging from PES-C-1 to PES-C-4. The aggregation of nanoparticles could be observed gradually and the morphology of fibers also became more heterogeneous in PES-C-4. The spinnability deteriorated with the increase in content of the colloidal SiO2 and resulted in more discernible liquid drops and beads distributed in fibers. EG and water as small molecules would disrupt and decrease the solubility of PES-C in DMAc which led to poor spinnability. We believed that there was a balance here when the amount of small molecules increased to a certain extent which would break the equilibrium and cause the PES-C insoluble and precipitate from the solution, this process was similar to film fabrication via phase inversion process. Different small molecules had different influence effect on this balance in which water was more remarkable than EG, that is, blending even a small amount of water (PES-C-5) with the spinning solution could greatly affect polymers solubility and spinnability. It could be seen that many fibers disappeared and became dense and homogeneous membranes (PES-C-5 and PES-C-6) after drying, so the colloidal SiO2 in water was not a suitable additive to fabricate the composite membranes.
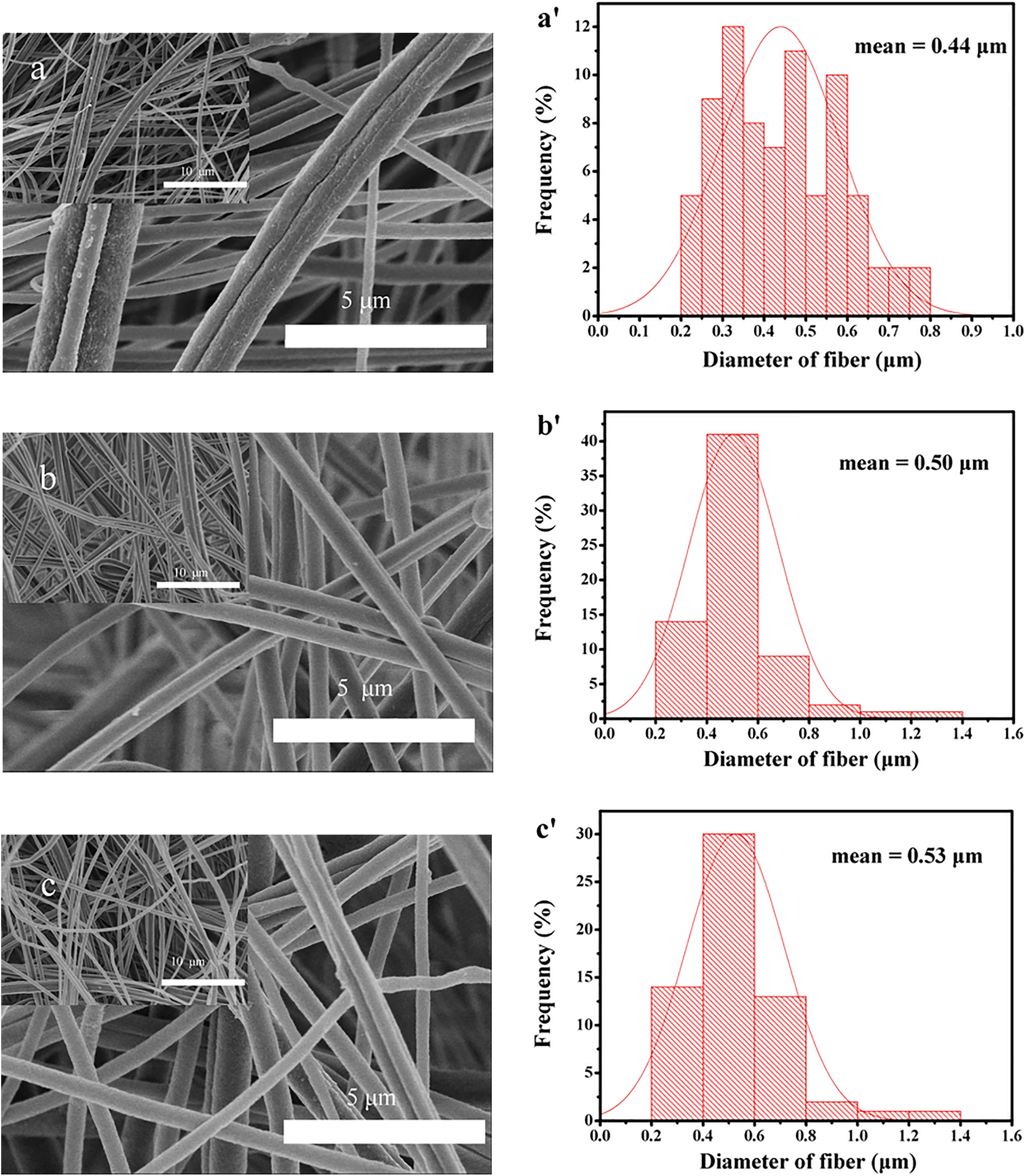
SEM images of PES-C and nanocomposite membranes: (a) PES-C-0, (b) PES-C-1, (c) PES-C-2, (d) PES-C-3, (e) PES-C-4, (f) PES-C-5, and (g) PES-C-6; corresponding fiber diameters: (a′) PES-C-0, (b′) PES-C-1, (c′) PES-C-2, (d′) PES-C-3, and (e′) PES-C-4. SEM: scanning electron microscopy; PES: phenolphthalein polyethersulfone.
Composition analysis of the nanofibers
The structure of the PES-C-0, PES-C-3, and PES-C-3 placed in air atmosphere for 10 days at room temperature were tested by FTIR spectra as shown in Figure 3. For the original PES-C membranes spectra (Figure 3(a)), the characteristic absorption bands at 1585 cm−1 due to the vibration of benzene ring and the peaks at 1324 cm−1 and 1150 cm−1 were associated with the stretching vibration of sulfone group S=O stretch. The peak at 1770 cm−1 was the C=O adsorption of phthalein cardo. When the SiO2 was added into the fibers, the characteristic adsorptions of PES-C-3 were similar with the spectra of PES-C-0, only the peak at 1107 cm−1 was reinforced which corresponds to the asymmetric stretching vibration of Si–O–Si, and the peak around 3500 cm−1 which was the characteristic absorption peak of –OH groups also seemed to be strengthened. When the PES-C-3 membrane was placed in the air for 10 days in the condition of about 60% relative air humidity, it could be seen that the intensity of the peak around 3200–3500 cm−1 increased obviously, which meant that the interaction between the membranes and moisture in the air was strengthened due to the incorporation of hydrophilic nanoparticles on the surface of the membranes.
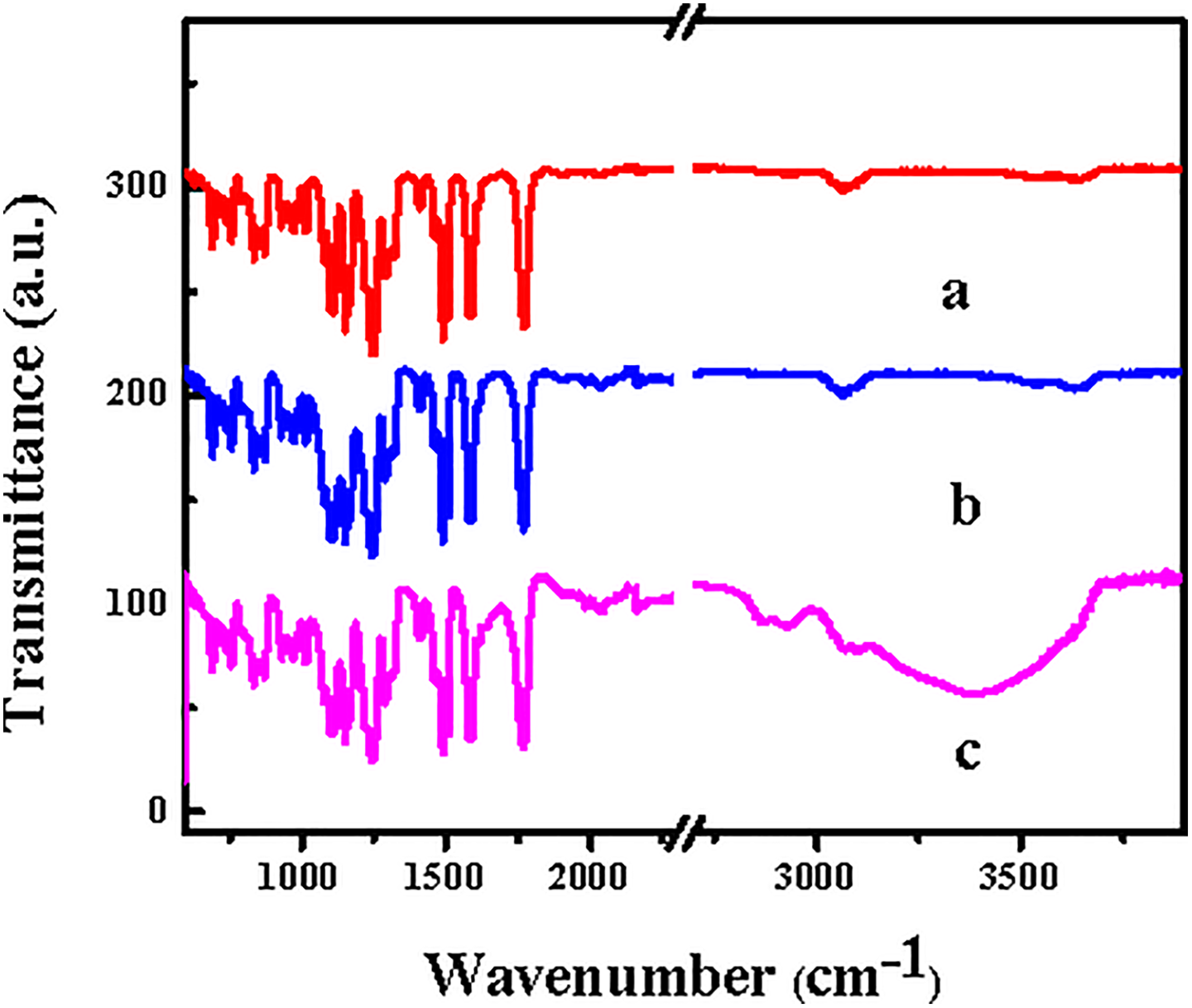
FTIR spectra of the (a) PES-C-0, (b) PES-C-3, and (c) PES-C-3 placed in the air for 10 days. FTIR: Fourier transform infrared; PES: phenolphthalein polyethersulfone.
The crystal structures of the nanofibrous membranes analyzed via WAXD are shown in Figure 4. The original PES-C membrane (PES-C-0) displayed completely amorphous diffraction patterns and showed only one major diffuse diffraction peak at about 18.89°. The main reason was the incorporation of phthalein group which disrupted the regularity of molecular chains and inhibited the close packing of the polymer chains. When the SiO2 was added into the spinning solution, the PES-C/SiO2 nanofibrous membrane was still amorphous and had two major diffraction peaks at 2θ were 20.70° and 29.28° (PES-C-3). With the increasing amount of SiO2, the nanoparticles began to agglomerate together and resulted in more diffraction peaks such as 16.80°, 29.42°, 31.04°, and so on, which meant the interactions between PES-C and SiO2 nanoparticles. In general, the structure of the composite membranes was amorphous while the nanoparticles dispersed well on the surface of the fibers.
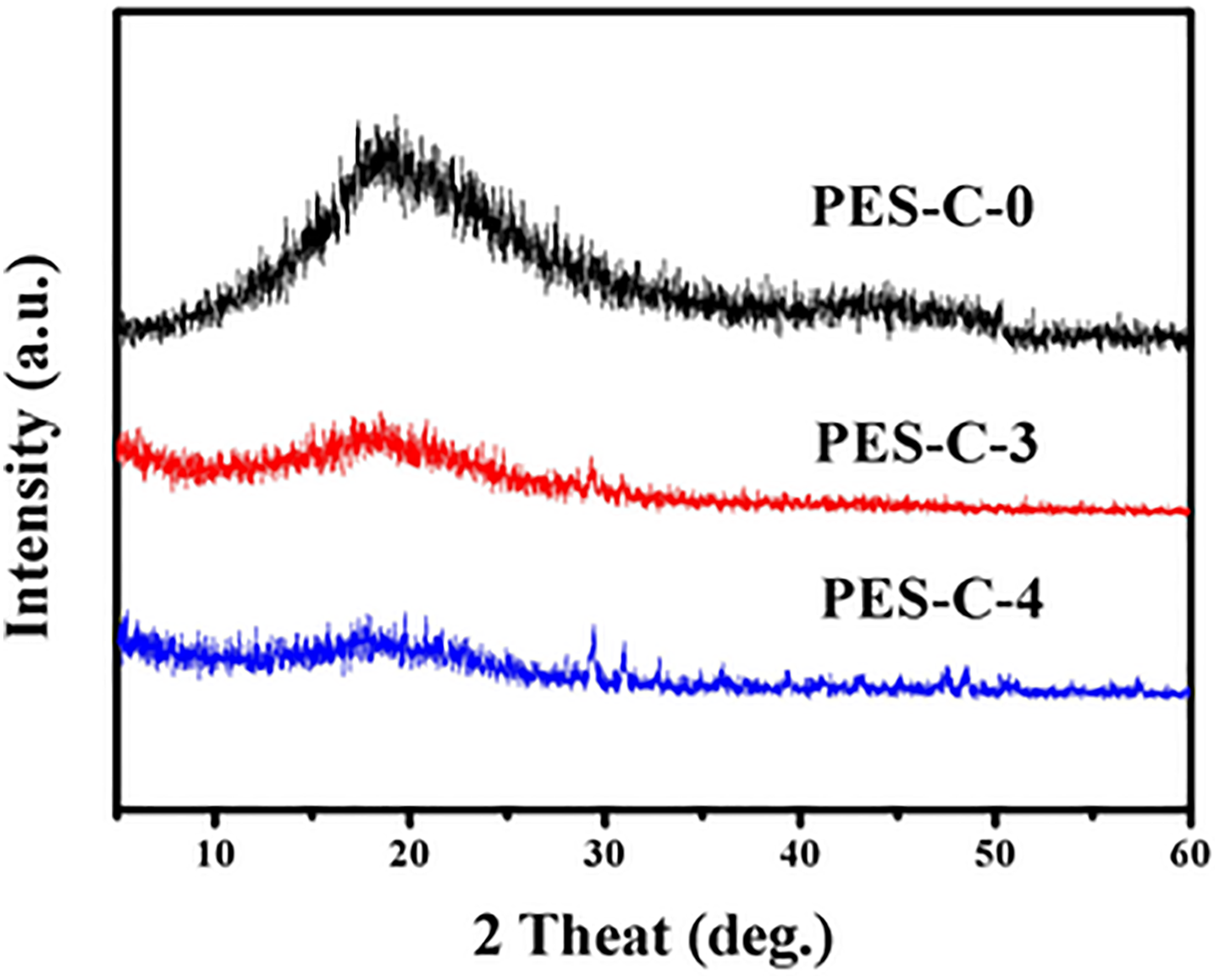
XRD spectra analysis of PES-C-0, PES-C-3, and PES-C-4. PES: phenolphthalein polyethersulfone. XRD: X-ray diffraction.
Thermal properties of membranes
The typical TGA curves of the membranes ranging from PES-C-0 to PES-C-4 are shown in Figure 5. It could be seen that all the membranes had excellent heat resistance performances and similar degradation mechanism of weight loss tendency. The weight loss below 200°C could be attributed to the escape of residual solvent and the hydroxyl groups on the surface of SiO2. The 5% weight loss temperature ranging from PES-C-0 to PES-C-4 was about 489.6°C, 490.6°C, 503.7°C, 497.0°C, and 493.1°C, respectively. With the addition of colloidal SiO2, it increased from 489.6°C to about 503.7°C at most corresponding to PES-C-2. But while the content of SiO2 continued to increase which resulted in not only the deterioration of polymers solubility and spinnability but also the aggregation of nanoparticles on the surface of the membranes, the thermal stability decreased again. Thermal decomposition temperature (Td) was similar at about 520°C which indicated the degradation mechanism of PES-C/SiO2 composite membranes that didn’t change with the incorporation of nanoparticles. The char yields of the membranes increased gradually from 47.96% to 52.93%. Overall, the incorporation of well-dispersion nano-SiO2 on the surface was helpful to enhance the thermal stability of the composite membranes.
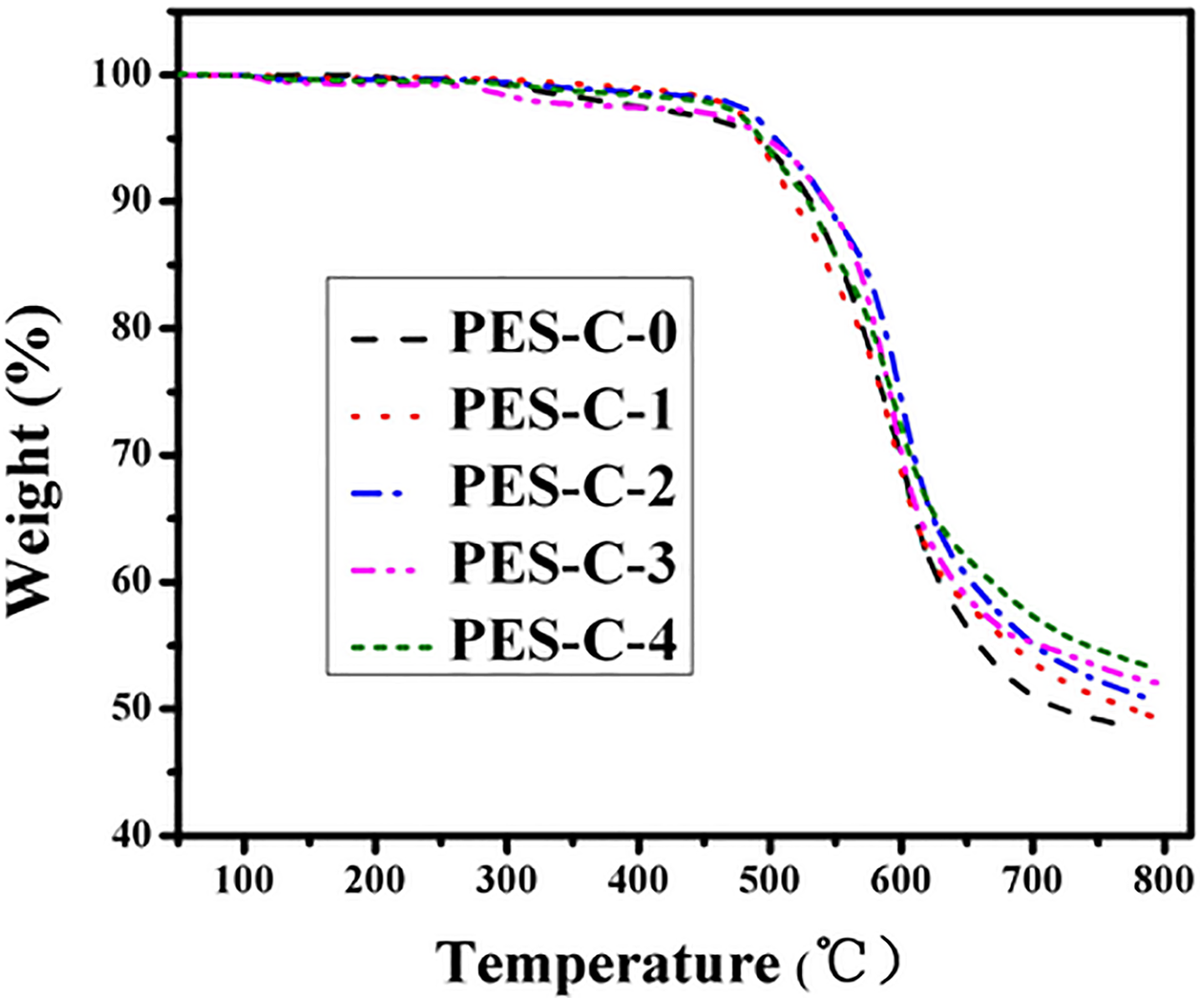
TGA curves of the nanofibrous membranes ranging from PES-C-0 to PES-C-4. TGA: thermal gravimetric analysis; PES: phenolphthalein polyethersulfone.
The surface wettability and porosity of the membranes
As we know that enhancing the hydrophilicity of the membranes could improve their antifouling ability during filtration process. Figure 6 shows WCAs ranging from PES-C-0 to PES-C-4 corresponding to about 135°, 131°, 123°, 115°, and 105°, respectively. The reason why WCAs decreased may be due to the following reason: First, the SiO2 nanoparticles which contained numerous hydroxyl groups could enhance the interaction between composite membranes and water; the second one was the decreasing of the membranes porosity. 26 But when the content of EG exceeded a certain limit in spinning solution, the aggregation of nanoparticles on the membranes’ surface could be observed in SEM images, which would increase surface roughness of the membranes, and it would lead to the enhancement of hydrophobicity. 20 –23 The porosity of the nascent PES-C membrane was about 85% due to its fluffy structure, and the parameter firstly increased and then decreased with the increase in the amount of the nanoparticles. In general, the porosity of the membranes would increase with the increasing fiber diameter; probably the reason for descending tendency of porosity might be because of more small beads and lumps that were sprayed on the membranes to block the gaps during the spinning process.
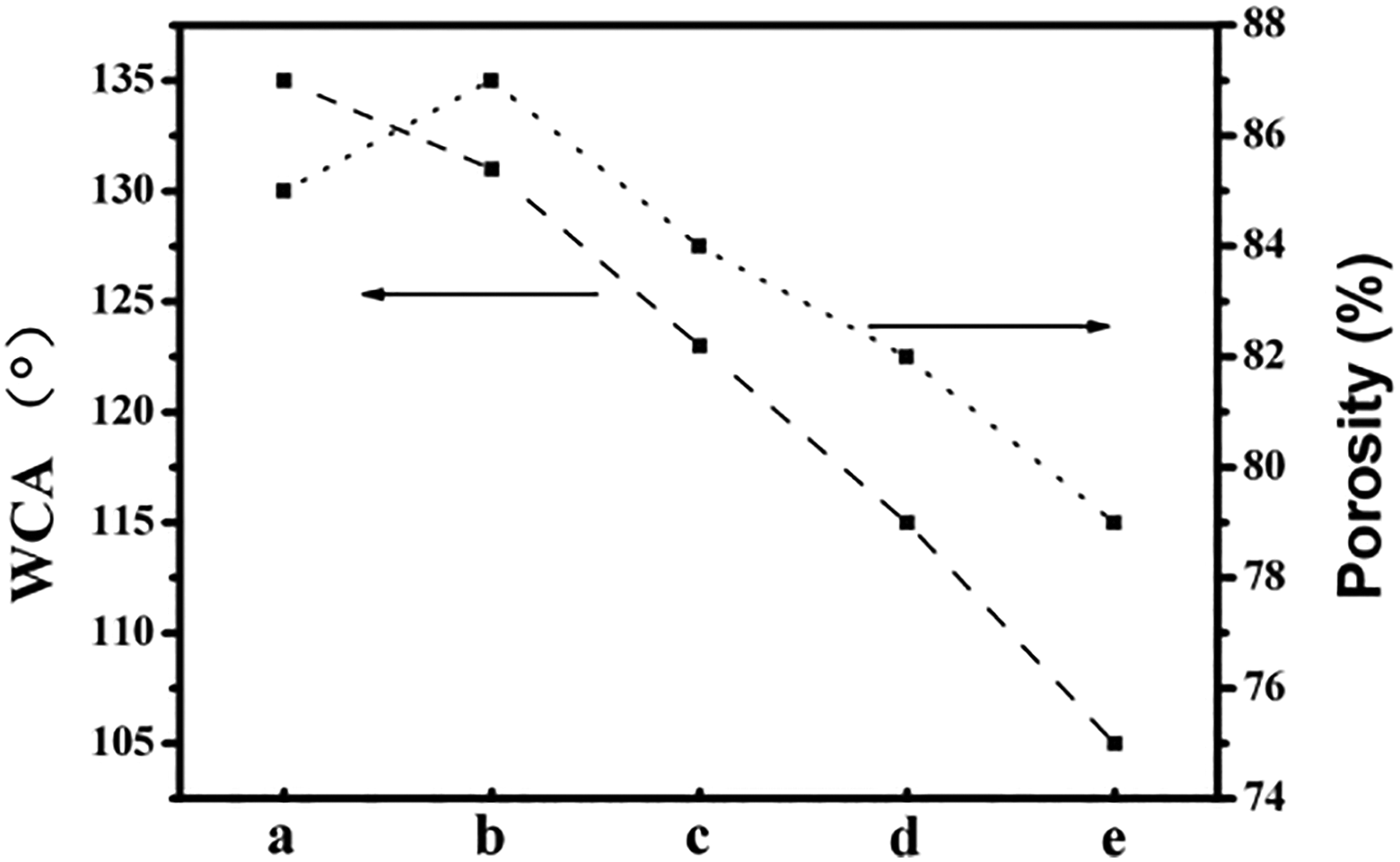
The WCAs and porosity of the original and composite membranes: (a) PES-C-0, (b) PES-C-1, (c) PES-C-2, (d) PES-C-3, and (e) PES-C-4. WCA: water contact angle; PES: phenolphthalein polyethersulfone.
Permeation and antifouling property of the membranes
Pore size of the membranes
Pore sizes were conducted by a CFP-1100-AI capillary flow porometer and the results are shown in Table 2. The mean pore size of original PES-C membrane was about 2.9959 µm. With the increase in the diameter of the original PES-C fibers, the composite membranes pore sizes increased slightly, but when the aggregation of nano-SiO2 exhibited seriously, the pore sizes decreased again maybe due to the clogging of the bulky particles.
Pore diameter of PES-C and PES-C/silica composite membranes.
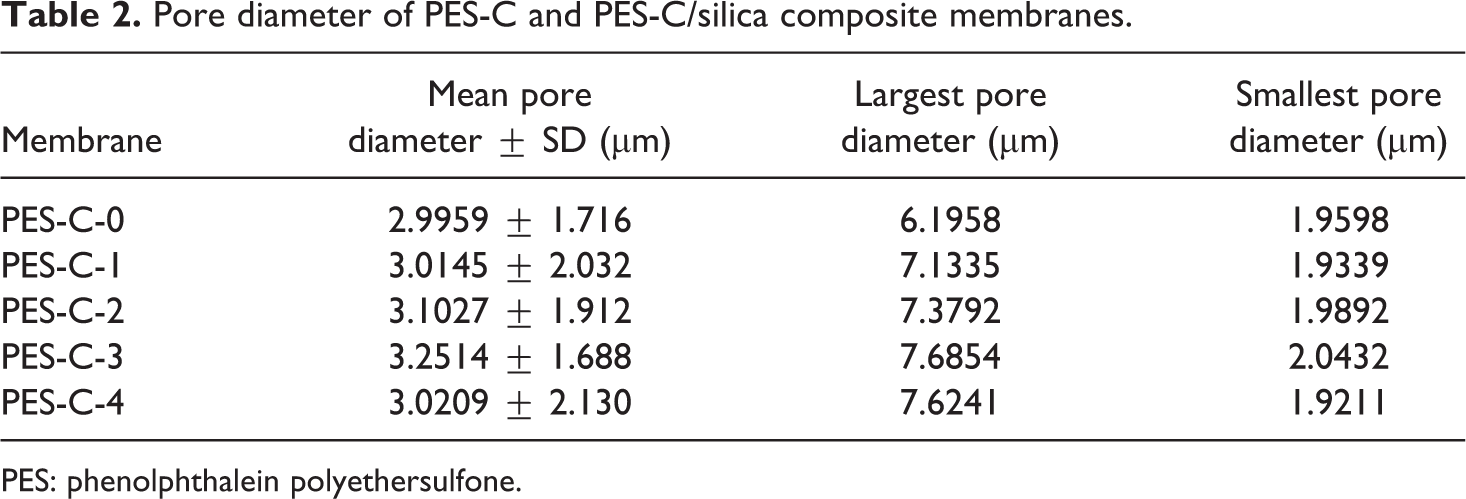
PES: phenolphthalein polyethersulfone.
The size of the starch particles is shown in Figure 7, which ranged from 1.7 to 2.6 µm with a volume average particle diameter of approximately 2 µm. The standard filtration media were suitable for analyzing the antifouling performances of composite membranes which the pore sizes belonged to the microfiltration field. 27,28 The concentration of the starch suspension was 2 g/L in the following antifouling experiments.
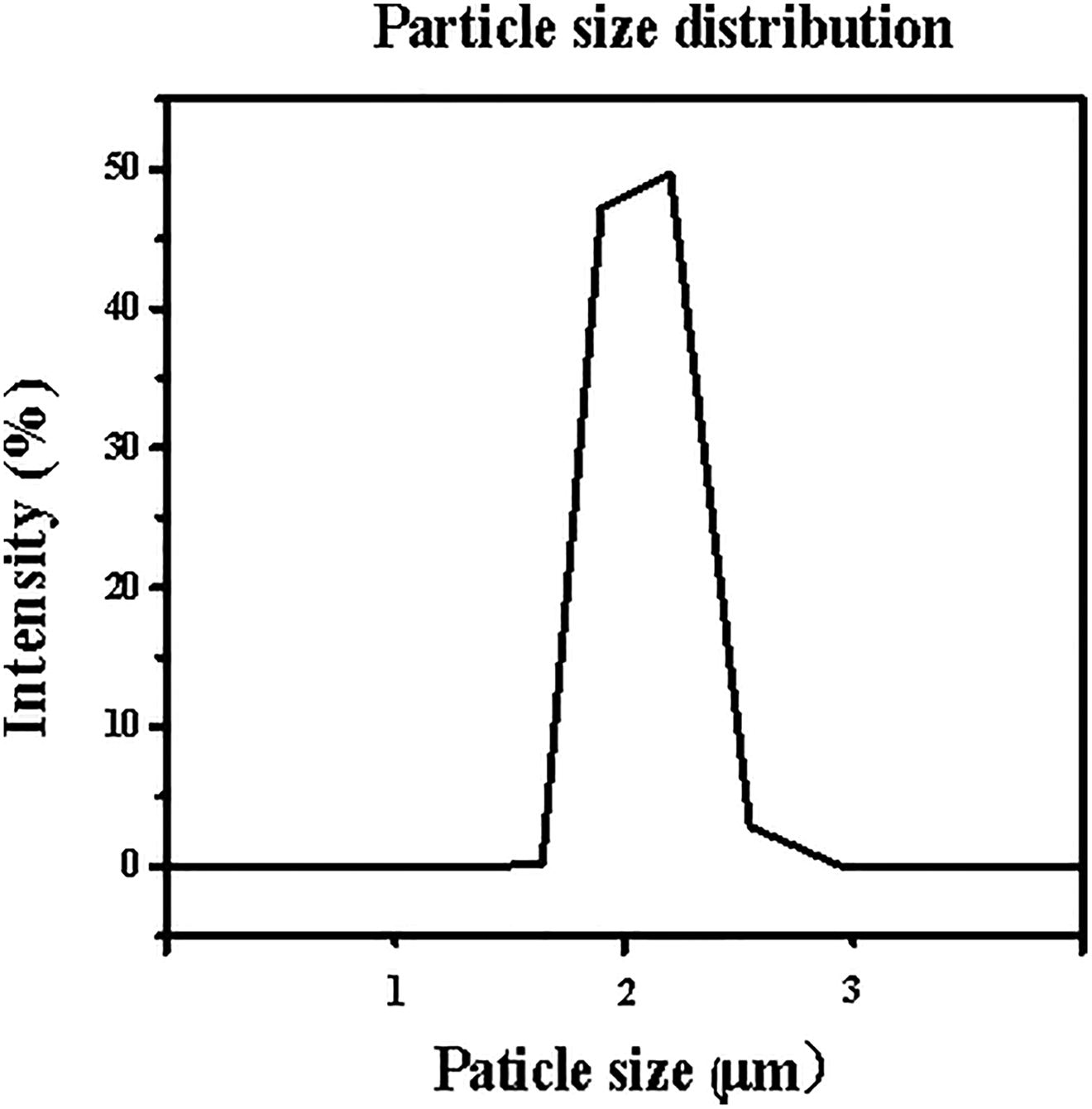
Particle size distribution of the starch particles.
A dead-end filtration system was designed to characterize the separation performance of the membranes. The water flux (Jw1) and recovery water flux (Jw2) of the membranes are depicted in Figure 8. The water flux of nascent PES-C membrane was about 6000 L/m2·h at 5 kPa; with the addition of SiO2, the water flux gradually increased to about 7000 L/m2·h corresponding to PES-C-3. After 60 min of the starch filtration under ambient pressure, the membranes were simply backwashed for 5 min, the recovery water flux (Jw2) of the membranes ranging from PES-C-0 to PES-C-4 was 3360 L/m2·h, 3600 L/m2·h, 3840 L/m2·h, 4900 L/m2·h, and 4800 L/m2·h, respectively, and the FRR were about 56.0%, 58.1%, 63.0%, 70.0%, and 72.7%, respectively. Although the hydrophilicity and FRR of PES-C-4 were slightly better than PES-C-3, polymers solubility and spinnability deteriorated that led to the worse of the fibers morphology. The reversible fouling was mainly created by starch cake on the membrane surface that was easily backwashed, and the irreversible fouling was caused by the starch attachment or adsorption on the surface and in the pores that was difficult to be cleaned up solely. 29 Therefore, the composite membranes exhibited better hydrophilicity to resist membranes’ fouling and the high water flux of the membranes could achieve fast and efficient separation in filtration pretreatment. In general, the PES-C-3 was comparatively more suitable to apply in the microfiltration field among these membranes.
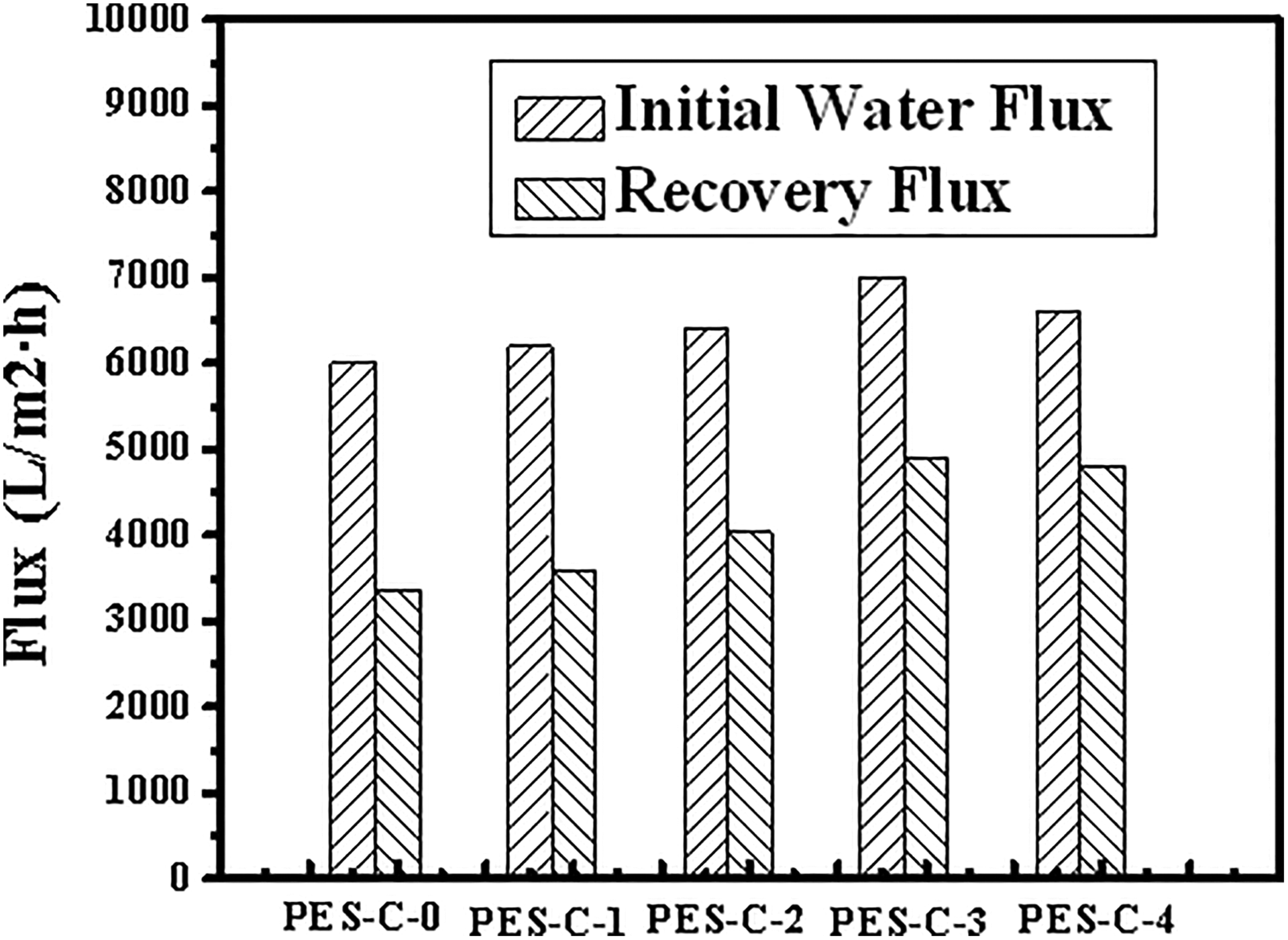
Water flux of initial water flux (Jw1) and recovery water flux (Jw2) at 5 kPa: (1) PES-C-0, (2) PES-C-3, and (3) PES-C-4. PES: phenolphthalein polyethersulfone.
Conclusions
PES-C/SiO2 composite membranes with various SiO2 contents were successfully fabricated via SB. The results showed that the nanoparticles dispersed well on the fibers, and the thermal properties were improved compared with pure PES-C membrane. The WCA measurements indicated that the hydrophilicity of composite membranes was enhanced, and the filtration experiments demonstrated that the antifouling property of the composite membrane was improved and it would be more suitable to apply in pretreatment of the filtration field.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This work was financially supported by the National Natural Science Foundation of China (no. 51473031) and Natural Science Foundation of Shanghai (no. 15ZR14011 00).