
Abstract
Bio-based high elastic polyurethanes were prepared from hexamethylene diisocyanate and various ratios of isosorbide to poly(tetramethylene glycol) as a diol by a simple one-shot bulk polymerization without a catalyst. Successful synthesis of the polyurethanes was confirmed by Fourier transform-infrared spectroscopy and 1H nuclear magnetic resonance. Thermal properties were determined by differential scanning calorimetry and thermogravimetric analysis. The glass transition temperature was −47.8℃. The test results showed that the poly(tetramethylene glycol)/isosorbide-based elastomer exhibited not only excellent stress–strain properties but also superior resilience to the existing polyether-based polyurethane elastomers. The static and dynamic properties of the polyether/isosorbide-based thermoplastic elastomer were more suitable for dynamic applications. Moreover, such rigid diols impart biocompatible and bioactive properties to thermoplastic polyurethane elastomers. Degradation tests performed at 37℃ in phosphate buffer solution showed a mass loss of 4–9% after 8 weeks, except for the polyurethane with the lowest isosorbide content, which showed an initial rapid weight loss. These polyurethanes offer significant promise due to soft, flexible and biocompatible properties for soft tissue augmentation and regeneration.
Introduction
The main biodegradable polymers used in biomedicine are polycaprolactone (PCL), polyglycolic acid (PGA), poly(L-lactic acid) (PLLA), and their derivatives.1,2 These polymers are all very stiff and are not suitable for tissue engineering of soft tissues, even in a fibrous or woven state. The development of softer, more flexible, and degradable polymers opens up a range of opportunities for soft tissue augmentation and regeneration.3,4
Polyurethanes (PUs) are a class of biodegradable polymers that have been applied for tissue-engineering scaffolds, as they have low cytotoxicity in vitro and in vivo.5–9 Moreover, plenty of nonbiodegradable PUs have been utilized in blood-contact applications such as for aortic grafts, dialysis membranes, breast implants, heart valves, and bone adhesives.10,11 In particular, segmented PUs are an important subclass of the family of thermoplastic elastomers, which consist of an alternating flexible component or macrodiol such as oligomeric PCL,12,13 poly(ethylene oxide),14,15 poly(lactic acid) 16 and poly(3-hydroxyburyrate diol) 17 called soft segment, and a stiff component derived from diisocyanate and a chain extender, called hard segment. 18 Soft segment is normally the amorphous state, which governs properties at low temperature and crystallizes only with a low level of hard segment content or prolonged cooling. The nature of hydrogen bonding in the hard segment causes a strong mutual attraction leading to formation of hard and soft segment domains. Hard domains act as thermo-reversible physical crosslinks and provide for the thermoplastic and elastomeric characteristics of the PU. 19
The choice of monomers used to synthesize biodegradable thermoplastic PUs is dependent on the final applications for the material. The properties of PUs are remarkably affected by the content, type, and molecular weight of the soft segments. 20 Mixed or special types of polyols are useful to impart specific properties. Several investigations have been carried out to clarify the effects of using different soft segments on the final PU properties.21–26 In contrast, hard segment is not limited to the structure of diisocyanate and the attraction through hydrogen bonding or molecular interactions. Bio-based bicyclic diol, isosorbide acts as a hard segment due to its structurally rigid properties.27,28
As interest in polymers for biomedical devices has increased, biocompatible and biodegradable polymer precursors have been used to synthesize segmented thermoplastic PU elastomers that can be visualized as possible candidates for many applications as vascular prostheses, soft tissue adhesives, pericardial patches, and in tissue engineering as cancellous bone substitutes.29,30
In this study, we present the synthesis of a new family of biocompatible and biodegradable PUs synthesized via simple catalyst-free, one-shot polymerization of hexamethylene diisocyanate, poly(tetramethylene glycol) (PTMG), and isosorbide. The absence of a catalyst has positive presumptions for toxicity. The chemical structure was confirmed by Fourier transform-infrared (FT-IR) spectroscopy and 1H nuclear magnetic resonance (NMR), and the physical properties were determined by differential scanning calorimeter (DSC), thermogravimetric analysis (TGA), and gel permeation chromatography (GPC). We also measured the mechanical properties and degradation rate, which generally correlated with the ratio of isosorbide to PTMG.
Materials and methods
Materials
PTMG 2000 was dried under reduced pressure at 60℃ for 4 h before use. Hexamethylene diisocyanate (HDI, Supplied by Aldrich, St. Louis, MO, USA) was used as received. 1,4:3,6-Dianhydro-D-sorbitol (Isosorbide, 98%) (Sigma-Aldrich, St. Louis, MO, USA) was used after drying at 40℃ for 6 h. Phosphate buffered saline (approximate pH 7.3) was obtained from Oxoid Ltd. (Basingstoke, Hampshire, England). N,N-Dimethylformamide (DMF), isopropanol and methanol were used without further purification.
Instruments
1H-NMR spectra for the synthesized PUs were recorded with a Bruker Avance 400 spectrometer at 400 MHz and performed at ambient temperature with 5% (w/v) polymer solution in CDCl3. Tetramethylsilane was used as the internal reference. FT-IR spectra were obtained using a Varian Australia 640-IR (Varian Australia Pty., Ltd, Sydney, Australia). A 2.5% solution of polymer in chloroform was placed directly onto a KBr pellet (Sigma–Aldrich, USA). Subsequent evaporation of chloroform at 50℃ under vacuum was performed for 2 h. The spectra did not show evidence of residual solvent. The weight average (Mw) and number average (Mn) molecular weights of the PUs were measured by GPC using a Futecs NP-4000 instrument (Futecs, Seoul, South Korea) equipped with a model P-4000 pump, a model AT-4000 column oven, GPC KF-804 column, and a Shodex (Shodex, Japan) R1-101 refractive index detector. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1.0 mL/min, and a sample concentration of 2.5 mg/mL was used. Polystyrene (Mw = 2000, 7000, 12,000, 65,000, and 120,000) was used as the standard. DSC data were recorded with a DSC (SEIKO Exstar 7020, Tokyo, Japan) instrument. Specimens (∼10 mg) were sealed in a DSC aluminum pan before being placed in the calorimeter, cooled to –70℃, and then heated to 300℃ at a rate of 10℃/min using a nitrogen atmosphere. TGA tests were conducted on the samples using Shimadzu TGA 50 (Shimadzu, Tokyo, Japan) equipment operating from 30℃ to 600℃ at a heating rate of 10℃/min and under a nitrogen atmosphere. Polymer films were prepared by solvent casting in DMF at a 10% polymer concentration followed by air drying to give films of 0.2 mm thickness. Tensile strength and elongation at break of the PUs were measured on an Instron universal testing machine (Model 3344, Instron Engineering Corp., Canton, MA, USA) at a crosshead speed of 10 mm/min at room temperature. The wetting ability of the polymer surface was evaluated based on contact angle measurements using PHX 300 contact angle equipment (S.E.O, Seoul, Korea). The surface energy calculation was carried out using the Girifalco–Good–Fowkes–Young method via PHX software. Scanning electron microscopic (SEM) photographs were obtained using a SEM (model JSM-5410LV, JEOL, Tokyo, Japan) at a magnification of 1000× or 1500×.
Preparation of polyurethane
Polyurethanes of different compositions with different content of isosorbide and PTMG and their yields.
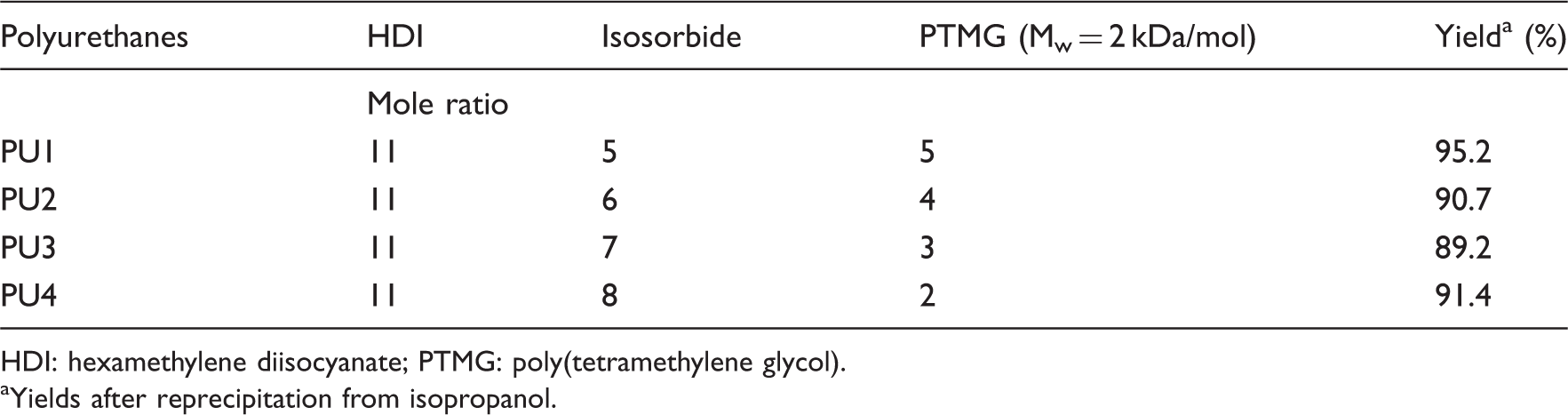
HDI: hexamethylene diisocyanate; PTMG: poly(tetramethylene glycol).
Yields after reprecipitation from isopropanol.
Degradation test
PU film (diameter, 25 mm) degradation was quantified by changes in dry weight. The PU samples were degraded for 2, 4, and 8 weeks. The dry films were weighed (W0) and immersed in a conical tube containing phosphate buffered solution (10 mL, approximate pH = 7.3). The degradation was conducted at 37 ± 1.5℃ in a water-bath (Daihan WiseBath, Seoul, Korea). Samples were taken at intervals, rinsed with water, dried in a vacuum oven for 2 days at 50℃, and weighed (Wt), after which they were discarded. The remaining weight was calculated as: Weight remaining (%) = W0 – Wt/W0
Cell culture
The PU films were sterilized by soaking them in 50%, 70%, and 100% ethanol for 30 min prior to use and then they were dried for 2 h. MC3T3-E1 mouse pre-osteoblastic cell line was maintained in standard T75 tissue culture flasks in normal growth medium composed of α-modified minimum essential medium (Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum (Gibco, Daejeon, South Korea) and 1% penicillin/streptomycin (Gibco). Prior to cell seeding, sections were cut from the synthesized PU films, placed into wells of 96-well plates, and seeded with 5 × 103 cells suspended in 1 mL normal growth medium and maintained at 37℃ in 5% CO2 for a subsequent time course analysis of cell number.
Cell proliferation
Cells were cultured on the PU films (the films were held down on the bottom of the plates with polytetrafluoroethylene insert rings) in 96-well plates for 1, 3, and 7 days. Cell proliferation was determined at these times using the Cell Counting kit-8 (CCK-8, 2 -(methoxy-4-nitrophenyl)-3 -(4-nitrophenyl)-5 -(2,4-disulfophenyl)-2H-tetrazolium monosodium salt) assay, according to the manufacturer’s instructions (Dojindo Laboratories, Kumamoto, Japan). Cell number was measured using the CCK-8 reagent. The CCK-8 solution was freshly prepared in growth medium, and 200 µL of CCK-8 reagent was added to each well, followed by incubation in a humidified atmosphere with 5% CO2. Absorbance was measured at 450 nm using a spectrophotometer (Bio-Rad, Seoul, South Korea). A blank experiment to detect cell-free background absorbance was also performed in parallel. Results are expressed as relative CCK-8 activity compared to control conditions.
Results and discussion
A family of aliphatic biodegradable and biocompatible PU elastomers based on hard segment isosorbide and soft segment PTMG were synthesized to design a number of implantable soft devices. The segmented PUs were composed of various ratios of soft segments to hard segments. Polymerization of HDI with various ratios of isorsorbide to PTMG was carried out by polyaddition-condensation at 120℃ for 12 h without catalyst, as shown in Figure 1. The ratios of isorsorbide to PTMG were 4:4, 5:3, 6:2, and 7:1. Bulk polymerization produced PUs of moderate molecular weight judging from the GPC data. The tough PU films were cast from a DMF solution.
Synthetic route and chemical structure of the polyurethane series.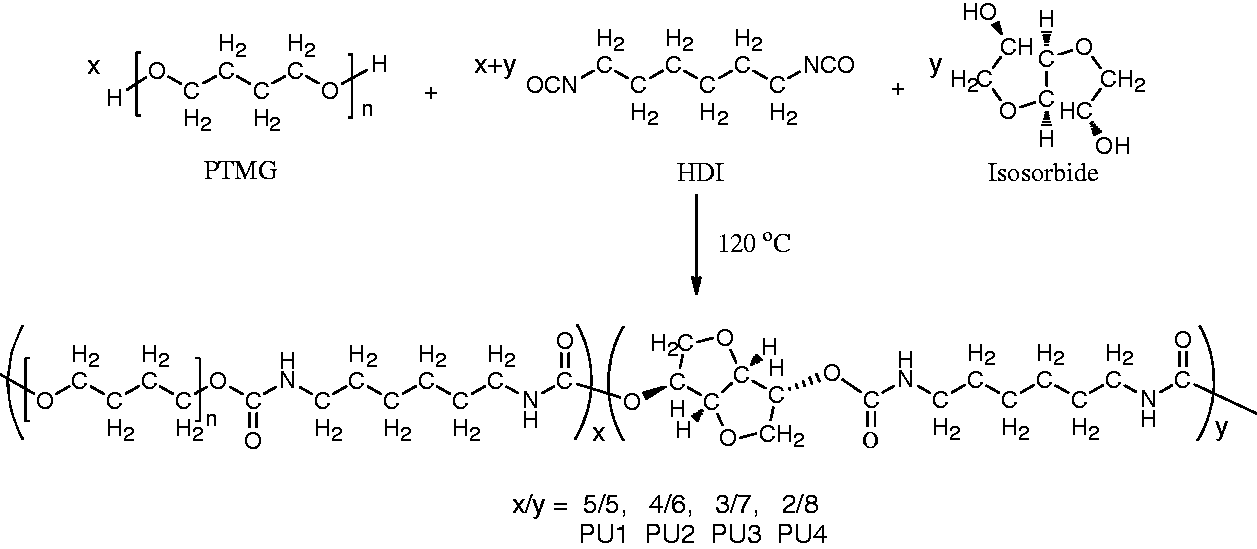
The FT-IR spectra of the PUs are shown in Figure 2(a). The PUs had a characteristic broad amide –NH– peak at 3325 cm−1 and strong carbonyl stretching absorption bands of amide at 1743 cm−1 and 1697 cm−1.
31
Hydrogen bonded NH bending bands were seen at 1535 cm−1. Asymmetric and symmetric CH2 stretching bands were seen at 2936 cm−1 and 2852 cm−1, 1108 cm−1, respectively. The disappearance of –N = C = O stretching vibration around 2250 cm−1 suggested that there was no unreacted isocyanate groups in any of the PU samples.
32
The IR spectra of PU1, PU2, PU3, and PU4 showed differences between the intensity of ethylene C–H stretching and ether combination absorption band around 2936 cm−1 and 2852 cm−1, and 1108 cm−1 indicating that the ratio of isosorbide to PTMG influenced the intensity of the C–H stretching band (Figure 2(b) and (c)).
Fourier transform-infrared (FT-IR) spectra of: (a) polyurethanes, isosorbide, PTMG and HDI; (b) C–H stretching; (c) C–O–C stretching bands of polyurethanes (PUs).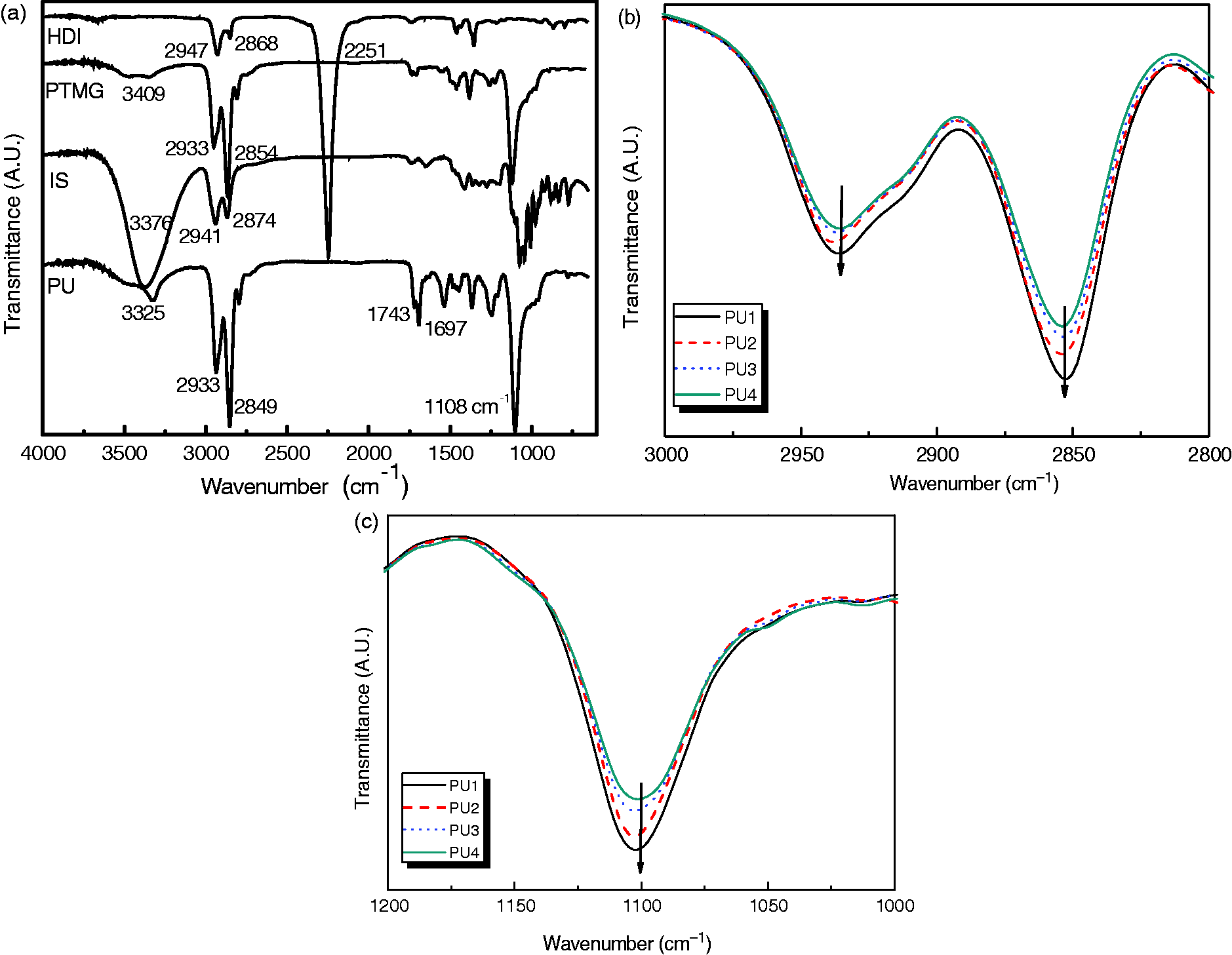
Figure 3 displays the
1
H NMR spectrum of the PUs, in which all proton signals of isosorbide, PTMG 2000, and the HDI segments were confirmed. The peaks occurring between 3.50 and 4.50 ppm were assigned to the bicyclic methylene protons of isosorbide. In addition, the peak at 8.07 ppm was assigned to the amine proton of the urethane N–H moiety. Signals occurring at 1.52, 1.64, and 3.18 ppm could be reasonably assigned to methylene protons of the HDI moiety.
1H Nuclear magnetic resonance (NMR) spectra of PTMG, isosorbide, HDI, and polyurethane.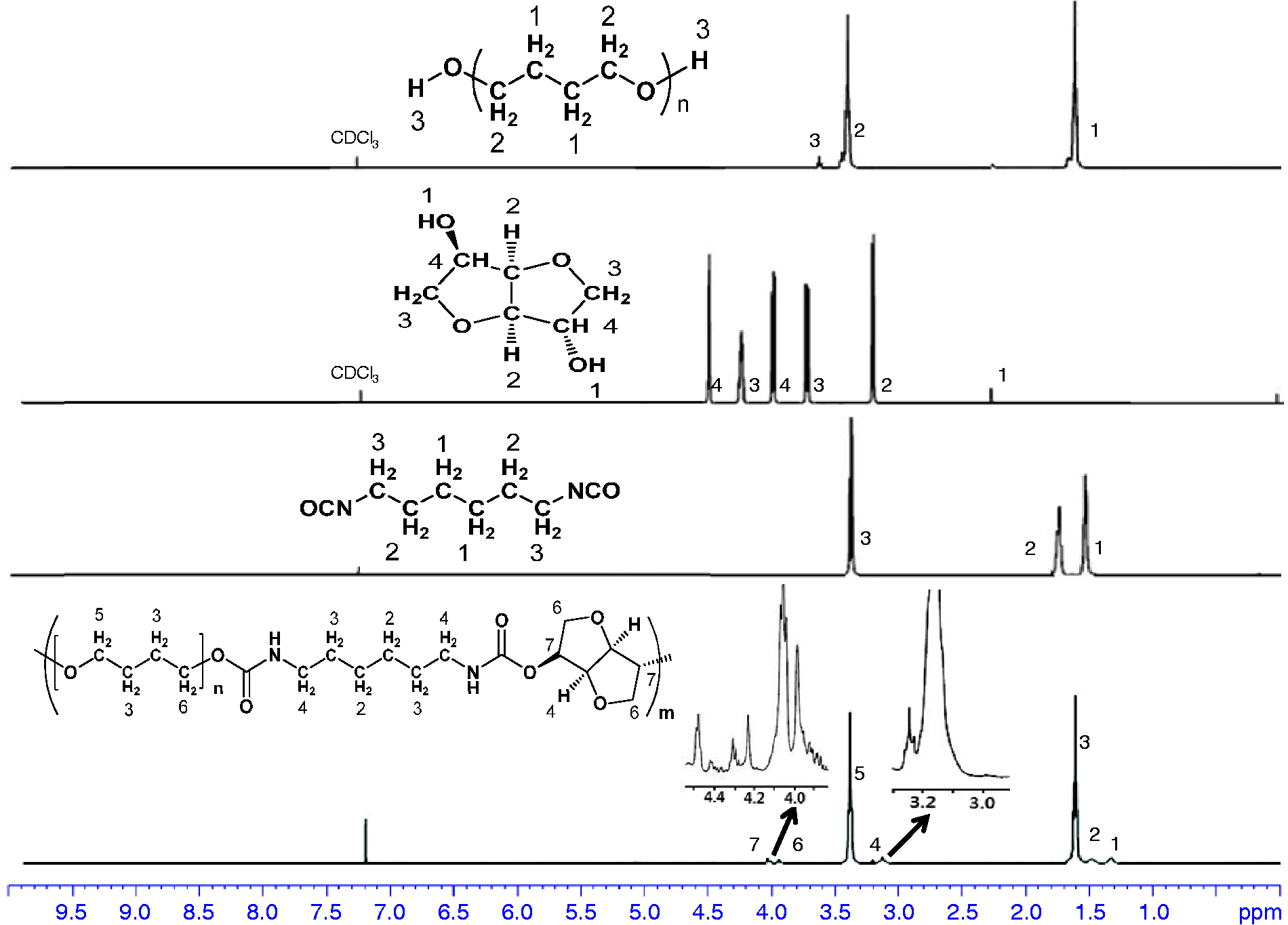
Molecular weights, thermal properties, and surface energy of the synthesized polyurethanes.
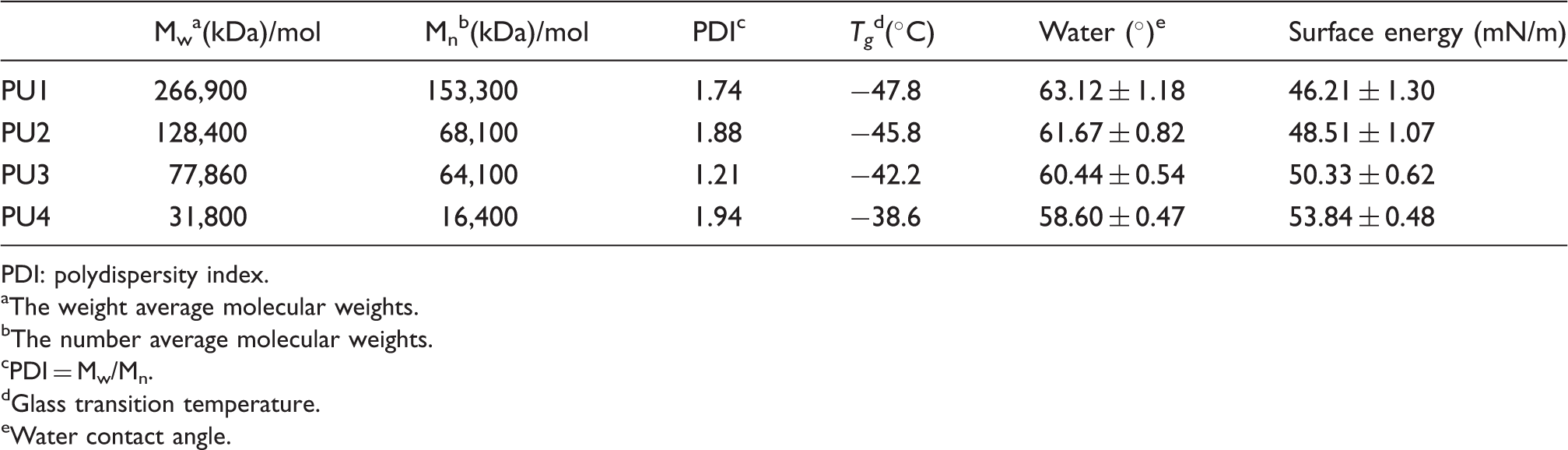
PDI: polydispersity index.
The weight average molecular weights.
The number average molecular weights.
PDI = Mw/Mn.
Glass transition temperature.
Water contact angle.
Much attention has been paid to the surface structures and properties of polymeric materials, because the functional groups on the surface play an important role in the interactions among materials with biological molecules and cells.14,15 As shown in Table 2, most of the PUs showed water contact angles < 65°. It is generally agreed that hydrophilic surfaces have contact angles with water in the range 1°–30° and those of hydrophobic surfaces are > 90°. Thus, the polymers presented here showed values somewhere between these ranges. The surface hydrophobicity of the PUs with different isosorbide content and PTMG2000 segments was characterized by static water contact angle measurements. As shown in Table 2, an increase in the isosorbide moiety in the PUs increased the contact angles slightly from 58.60° to 63.12°, which improved the hydrophobic nature of the polymer surfaces.
The stability of the polymer films under physiological conditions is very important for biomedical applications, because an unstable polymer can induce undesirable or unexpected loss of biological function and activity of the biomaterials.
22
For example, if the glass transition temperature (Tg) of the polymer is above that of body temperature, the polymer is in a rubbery state.
23
Table 2 shows the data collected from DSC measurements of the four PUs. The DSC analysis was conducted to characterize the thermal behaviors of the PUs. The endothermic transition at around 20℃ is melting transition of crystallized soft segments.
24
Tg was detected for all samples, as shown in Figure 4(a). The Tgs of PU1, PU2, PU3, and PU4 were −47.8, −45.8, −42.2, and −38.6℃, respectively, indicating that all PUs showed high elasticity even at low temperature. The resistance to heat of the PUs was examined by TGA, as shown in Figure 4(b). All PUs showed two-stage decomposition in the TGA thermograms, which was due to the existence of flexible alkylene and bicyclic units. It can be seen that the temperatures of 10% weight loss of the PUs decreased from 340℃ to 299℃ as isosorbide content increased, suggesting that all PUs were thermally stable and could tolerate sterilization.
(a) DSC and (b) TGA thermograms of polyurethanes: PU1, PU2, PU3, and PU4.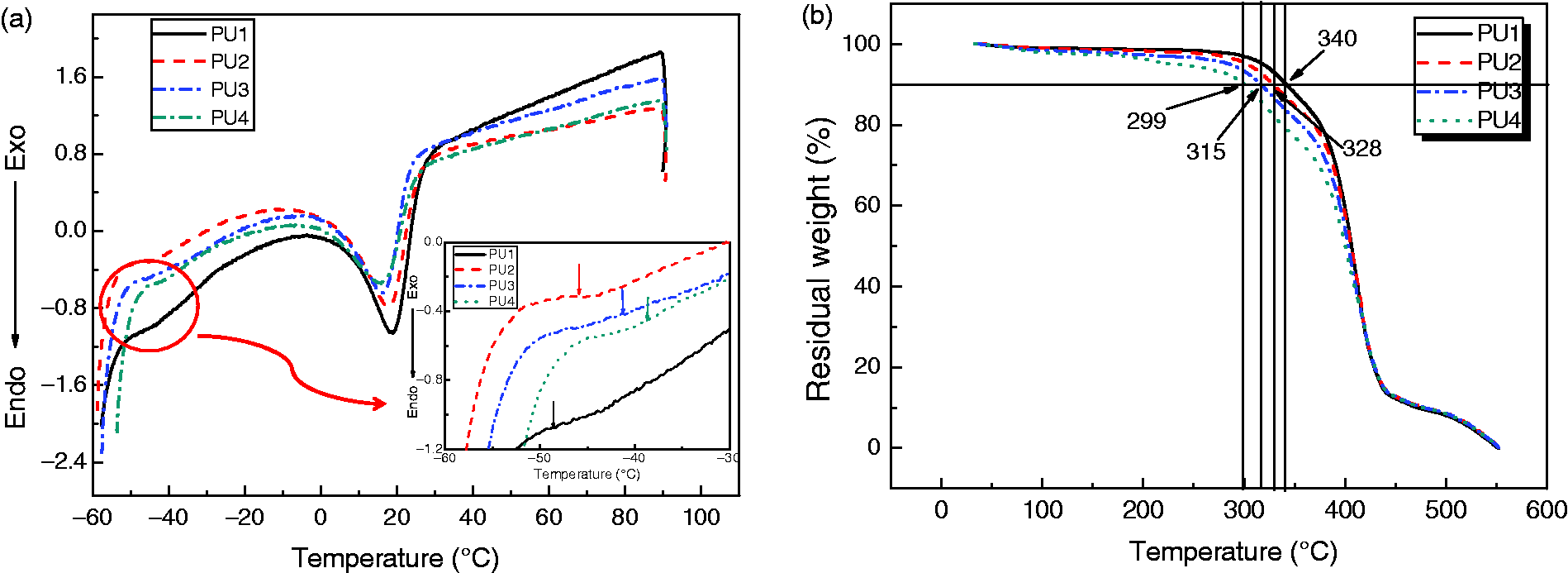
Figure 5 shows ultimate tensile strength, mean stiffness, and breaking strains for the PU samples. The PU films were flexible with ultimate tensile strengths (UTS) from 5.95 ± 0.35 MPa to 19.12 ± 1.25 MPa, mean stiffness (Young’s modulus) from 16.12 ± 0.64 MPa to 41.5 ± 2.12 MPa, and breaking strains from 153% to 1695% for PU1 and PU4, respectively. The UTS of the PU1 film was comparable to that of the aorta (50–100%),
34
whereas the breaking strain was superior to that of the aorta. This is the first example of a high breaking strain of 1600% in an elastic biopolymer. Furthermore, the UTS value was retained at 19.12 ± 1.25 MPa at that point.
(a) Ultimate tensile strength (UTS); (b) Young’s modulus; (c) strain at breaking (%); (d) example of tensile stress–strain curves for the polyurethane (PU) series.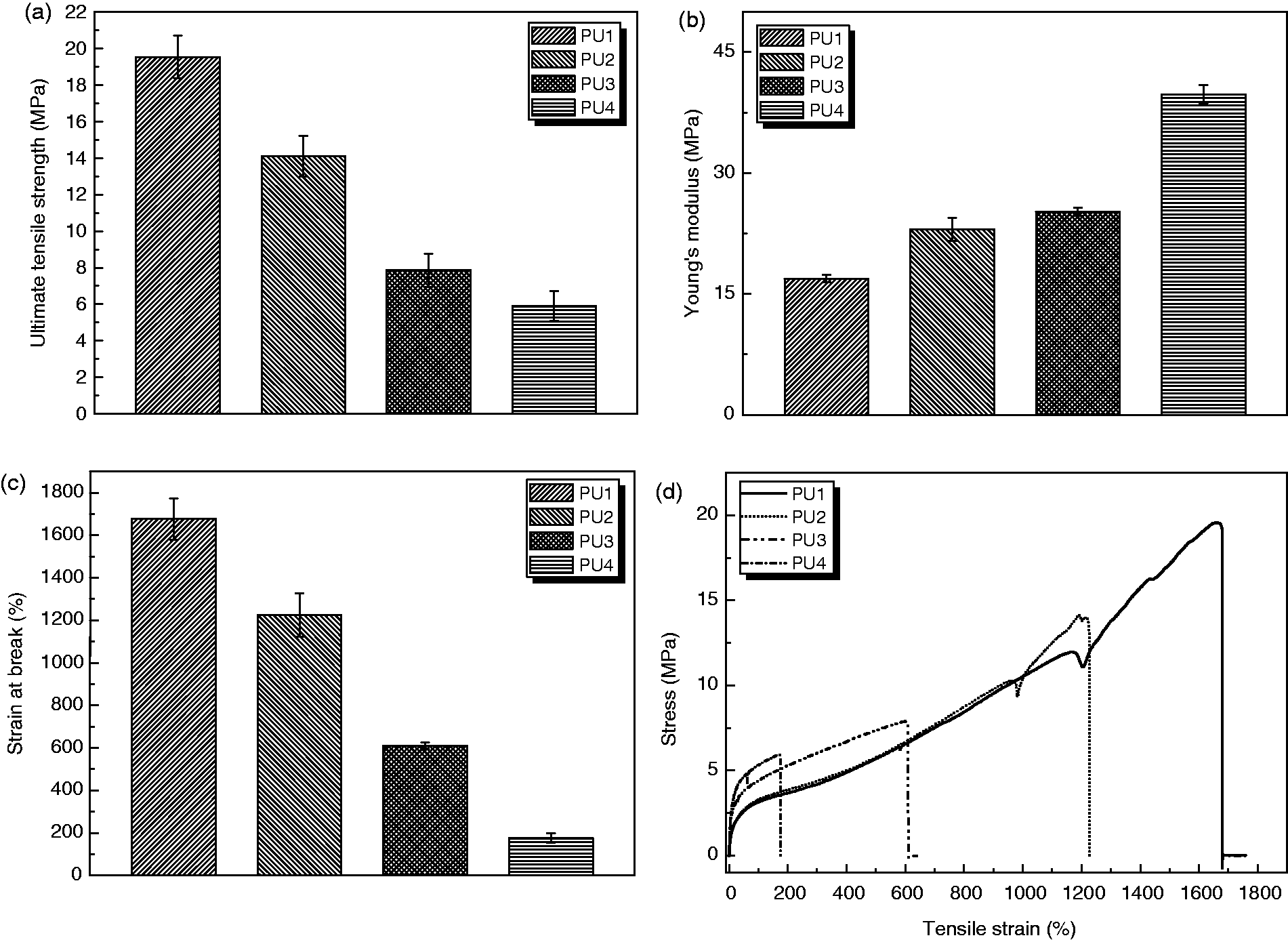
There was a relationship between Mw and UTS with increasing UTS and increasing Mw. In addition, stiffness increased with increasing isosorbide (hard segment).35,36 The main degradable polymers used in biomedicine are PCL, PGA, and PLLA and their derivatives. These polymers are all very stiff and are not suitable for utilization in tissue engineering applications for soft tissues, even in a fibrous or woven state. The development of softer, more flexible, and degradable polymers, as shown in this study, opens up a range of opportunities for soft tissue augmentation and regeneration, and the polymer family synthesized in this study offers such properties.
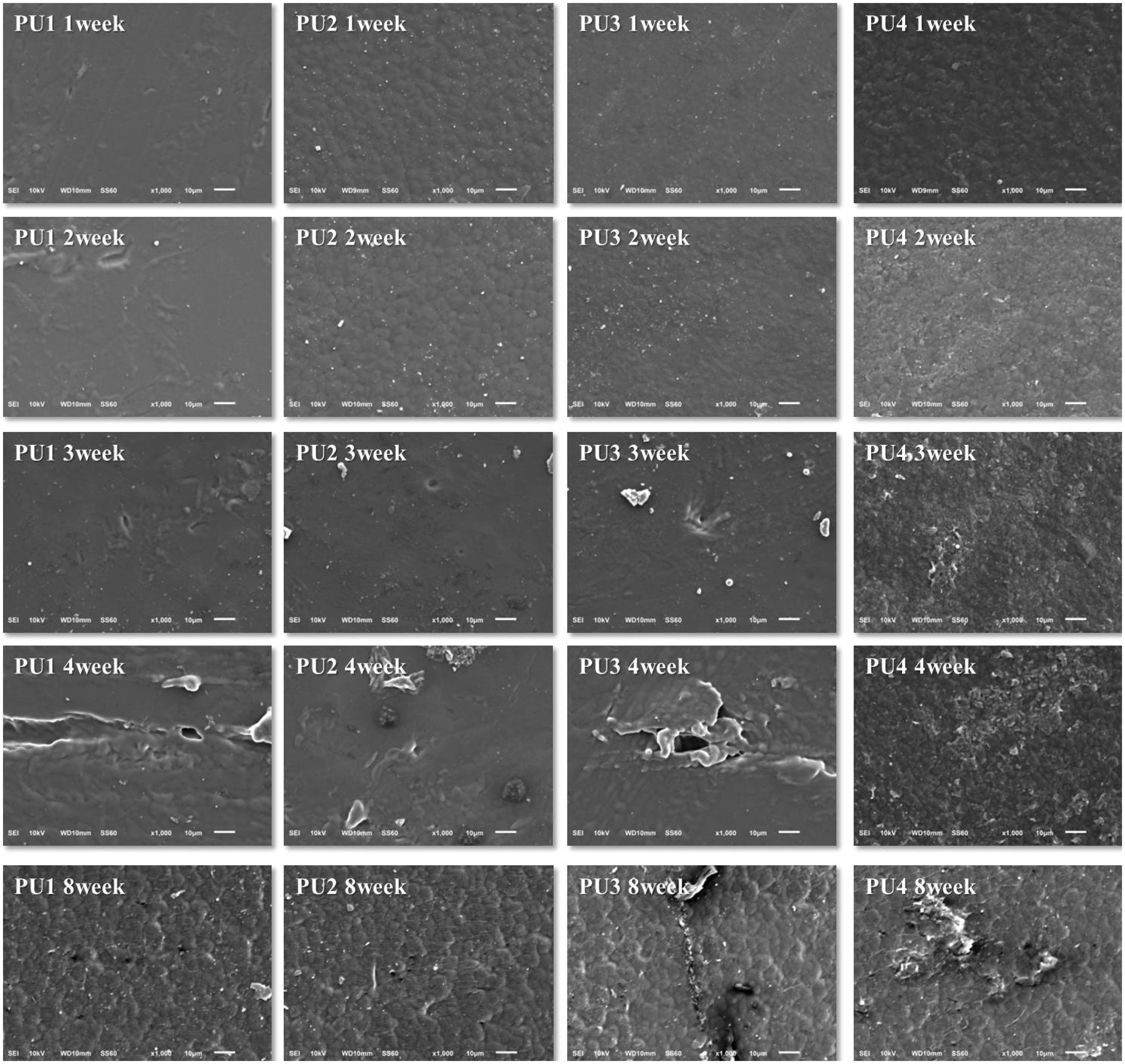
The degradation behavior of the PUs was examined in vitro. Samples were immersed in a phosphate buffer solution at 37℃. Figure 6 shows the percentage weight loss of the samples with time. Between weeks 0 and 1, PU1, PU2, and PU3 lost 2–3% of their weight, whereas PU4 showed a loss of about 4%. A continual slow weight loss was observed for PU1, PU2, and PU3 at the end of the test, and the final weight lost over 8 weeks was about 3%. However, the rate of PU4 weight loss was much faster than that of PU1, PU2, and PU3 over the 8 weeks, indicating that degradation of PUs with high molecular weight is slower than that of the low molecular weight PUs. In addition, the highly hydrophobic PU1 had a slower rate of water diffusion than those of the other PUs with higher isosorbide content. The surface of all polymers became rougher with time, although the extent of change differed depending on the composition of PUs tested. SEM photographs showed the changes in film surface after degradation. All surfaces initially appeared relatively smooth with few defects. However, the surface of all samples became rougher with time (Figure 7) and was significantly higher for PU4. The presence of isosorbide imparted a stronger hydrophilicity to the PU segment than did PTMG, which increased the rate of hydrolytic degradation.
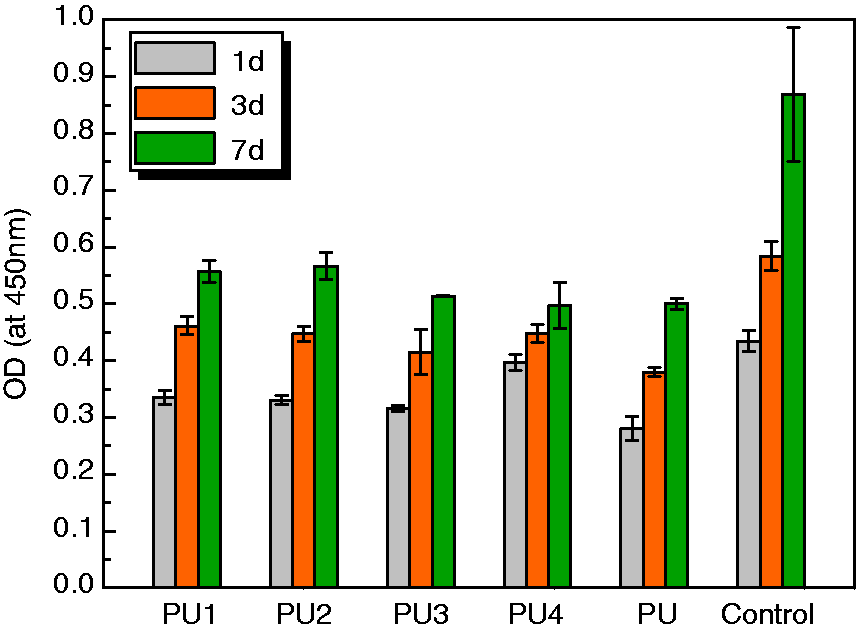
Cell attachment was examined with the MTS assay. The ODs at 450 nm for PU4 were 0.18, 0.25 and 0.32 after 1 -, 3 -, and 7-day cultures, indicating that the number of cells on the films had increased significantly. The results of CCK-8 absorbance values for MC3T3-E1 cells adhesion to and proliferation on the PU films compared to that of control are shown in Figure 8. It reflects not only the ability of the PU film to promote cell adhesion and proliferation, but also its cytotoxicity. Lower cell viability was observed from 1 to 7 days on the PU surfaces in comparison with that of the control.
Residual weight (%) of the polyurethane (PU) films after in vitro degradation in phosphate buffer solution: (a) PU1, (b) PU2, (c) PU3, and PU4. Scanning electron micrographs of hydrolytic degraded polyurethanes in phosphate buffer solution during 8 weeks: (a) PU1, (b) PU2, (c) PU3, and PU4. CCK assay of mouse MC3T3-E1 cultured on polyurethanes. PU: polyurethane obtained from PTMG and HDI.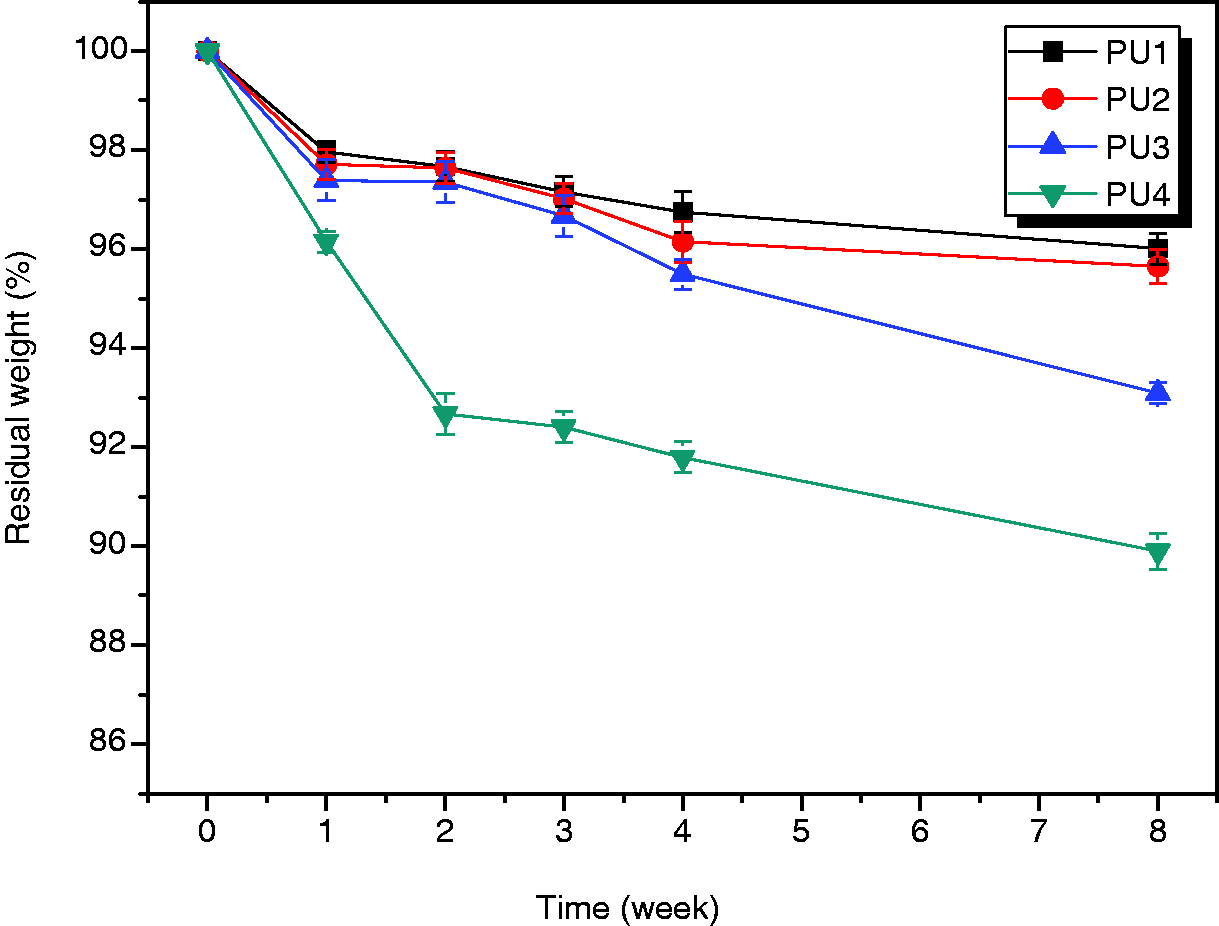
The MC3T3-E1 cells were examined by SEM after they were cultured on the PU films for 1, 3, and 7 days. The SEM images of the MC3T3-E1 cell morphology after being cultured on PU film for 7 days are shown in Figure 9. The SEM micrographs revealed that the cell appeared to spread and occupy the PU film surface. In most areas, cell density appeared to be relatively high with close contact. Hence, these findings show that our PU films had no negative effect on cell attachment or proliferation. Proliferation was assessed by the MTS assay and revealed that our PUs were biocompatible and nontoxic. In addition, results of the MTS assay suggested that our composite scaffolds exhibited an appropriate environment for the loaded cells, which appeared to proliferate well during the cultivation periods. But viability of PU4 is less than 50% of the control at day 7. The low molecular weight or oligomeric product would act as a big variable in determining low viability of PU4.
Scanning electron microscopic images illustrating morphology of MC3T3-E1 cells after being cultured on polyurethane film at day 7: (a) PU1; (b) PU2; (c) PU3; (d) PU4.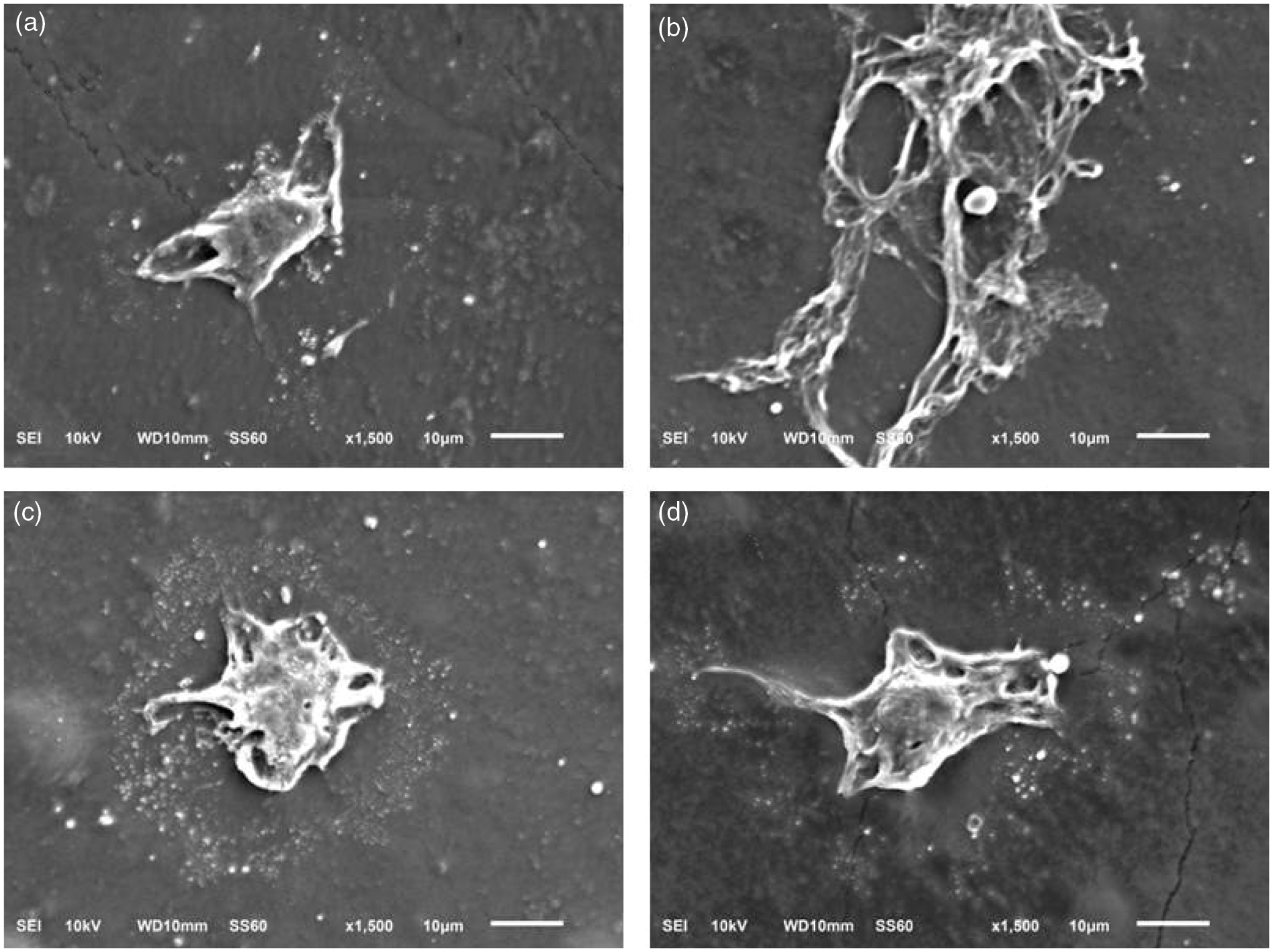
Conclusion
We introduced high elastic thermoplastic PUs which were synthesized by a simple one-shot polymerization of HDI, a hard segment of isosorbide, and a soft segment of PTMG as the diol. The PU1 had good mechanical properties with 1600% tensile strain at 18 MPa tensile stress. MC3T3-E1 cells from the calvaria of a C57BL/6 mouse embryo/fetus attached, grew, and proliferated favorably on these PU films. These findings suggest that the high elastic thermoplastic PUs obtained biomimetically have high potential to be used as a tissue engineering scaffold and for other biomedical uses.
Footnotes
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
This study was conducted by the research fund of Dankook University in 2013. This study was supported by Priority Research Centers Program (no. 2009-0093829) through the National Research Foundation of Korea.