
Abstract
Understanding the health consequences of chronic disruption of circadian rhythms can contribute to improving prevention strategies for shift workers. Chronic circadian disruption in shift work has been linked to a higher risk of stroke. Dysregulated immune responses are also linked to circadian disruption and may be a factor in stroke outcomes in shift workers. In this study, we test the hypotheses that specific schedules of circadian disruption exacerbate inflammatory responses in the brain, causing an increase in infarct size after experimentally induced ischemic stroke. Mice were exposed to 1 of 5 different lighting schedules followed by a 30-min middle cerebral artery occlusion, then reperfusion and 3-day recovery. A history of weekly phase advances resulted in an increased infarct volume versus the control lighting schedule. These effects were shift-direction specific, nonpermanent, and required multiple shifts to occur. In a separate cohort, stereotaxic injections of lipopolysaccharide were given bilaterally after exposure to 1 of 3 different lighting schedules. Ratios of pro- to anti-inflammatory cytokine expression show dysregulated responses after a history of phase advances. We conclude that chronic circadian disruption leads to worsened stroke outcome in a direction- and schedule-specific manner likely because of priming of the inflammatory response in the brain. These pieces of evidence suggest that the health impacts of shift work may be improved by targeting shift work scheduling, inflammatory mediators, or both.
Keywords
Twenty percent of the U.S. labor force work nontraditional hours and have a higher incidence of chronic health disorders, including cancer, obesity, diabetes, and cardiovascular disease (Tenkanen et al., 1998; Schernhammer et al., 2003; Karlsson et al., 2005; Wang and Namura, 2011). In addition, the Nurses’ Health Study has revealed an increased risk of vascular disease correlated to the length of a career that included rotating shift work (Brown et al., 2009). Disruption of circadian rhythms by shift-work schedules may play a mechanistic role. Circadian rhythms are endogenous oscillations generated at a cellular level with a period of approximately 24 h that regulate the timing of neuronal, metabolic, and behavioral output. These rhythms can be entrained by external cues such as light and dark cycles (Vitaterna et al., 2001). When circadian rhythmicity is altered by constant changes in the environment, such as nighttime light exposure, it can be disruptive to normal timing relationships among the environment, the brain, and the body (Yamazaki et al., 2000; Morris et al., 2016; West et al., 2017b).
Recently, rodent models of environmental circadian disruption (ECD) have been employed to establish causality and to uncover mechanisms of the relationship between circadian disruption and stroke risk. A recent study from Earnest and colleagues (2016) revealed a dramatically increased rate of mortality in male rats from surgically induced ischemic stroke after ECD. Females in that study were protected from mortality but exhibited a near doubling of infarct volume. However, another group (Ku Mohd Noor et al., 2017) did not observe such effects after permanent middle cerebral artery occlusion (MCAO). Thus, the association between shift work and stroke risk is suggested but not certain, and the exact cause remains unknown.
The immune system exhibits circadian rhythmicity. Isolated immune cells exhibit rhythms in response to endotoxins (Boivin et al., 2003; Keller et al., 2009; Bollinger et al., 2011). Rest and activity cycles determine the oscillation of hematopoietic cells, hormones, and cytokines in diurnal and nocturnal species (Scheiermann et al., 2013). ECD has been shown to reset clocks in immune cells and thought to play an important role in the deleterious health consequences of shift work in humans (Cuesta et al., 2016). Mice experience increased mortality (Halberg, 1975) or heightened inflammatory responses (Adams et al., 2013) after immune challenge in the rest period versus the active period. In vivo ECD followed by in vivo or in vitro immune challenge led to heightened mortality and inflammatory signaling (Castanon-Cervantes et al., 2010; Adams et al., 2013). Such effects also occur after in vitro ECD in isolated spleen explants (Stowie et al., 2019).
Dysregulated inflammatory signaling during and after circadian disruption may provide a mechanism for why stroke risk/severity increases due to shift work conditions. Neuroinflammation after stroke contributes to the severity of the resulting neurological deficit (Chapman et al., 2009; Dugue, 2017). Lipopolysaccharide (LPS)–induced inflammation before MCAO was shown to exacerbate infarct volume in mice and increase neuroinflammation (Doll et al., 2015). Indeed, the innate and adaptive immune response in the central nervous system (CNS) after ischemic stroke is predictive of complications during recovery (Iadecola and Alexander, 2001; Prass et al., 2003; Shi et al., 2018). In this study, we sought to more fully investigate innate immune responses in the brain as a causal link between ECD and the severity of brain tissue injury after ischemic stroke. We report that stroke severity is increased in mice after ECD in a schedule-dependent manner, potentially because the CNS becomes primed for a dysregulated immune response, resulting in increased brain damage.
Materials and Methods
Mice
Fifteen-week old male C57BL/6J mice were purchased from the Jackson Laboratory (product 000664), individually housed, fed ad libitum, and randomly assigned to 1 of 5 schedule conditions (Table 1): group A, control mice in which the light cycle was a standard, 12:12 LD (light:dark) schedule; group B, 4, once-per-week 6-h phase advances of the light cycle followed by 1 week of a static 12:12 schedule; group C, 4, once-per-week 6-h phase advances followed by 3 weeks of static LD; group D, 4, once-per-week 6-h phase delays followed by 1 week static; and group E, one 6-h phase advance followed by 1 week static. Here, the term static refers to a period between the last shift of the light schedule and the stroke-inducing surgery under a 12:12 LD cycle. All animal procedures used in this study were conducted in compliance with the National Institutes of Health, Guide for the Care and Use of Laboratory Animals, and were approved by the Morehouse School of Medicine Institutional Animal Care and Use Committee.
Light schedules employed for infarct study.
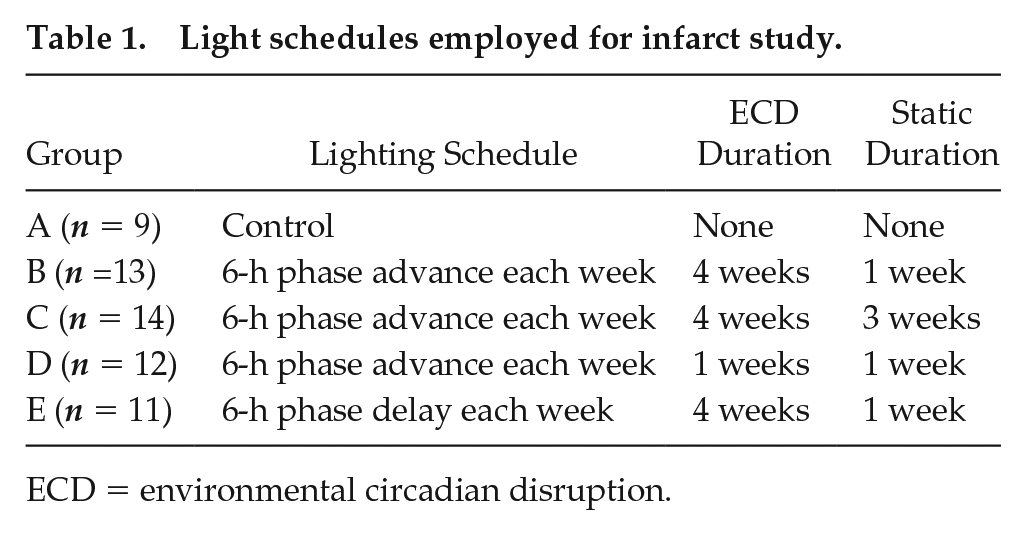
ECD = environmental circadian disruption.
Transient MCAO
To induce a mild ischemic stroke, a surgeon, masked to the experimental groups, performed a filamentous MCAO as previously described (Steele et al., 2008). The surgery was performed 1 week after the last lighting manipulation, between 0900 and 1200 h (ZT 2–ZT 5). Mice were anesthetized with 1.5% isoflurane in 68.5% N2O and 30% O2. MCAO was induced using a nylon monofilament that was inserted into the external carotid artery and advanced through the internal carotid artery to the origin of the middle cerebral artery. The filament was left for 30 min and removed to allow reperfusion. Changes in regional cerebral blood flow (rCBF) during MCAO were measured using laser-Doppler flowmeter (FLO-C1, Omega Wave, Tokyo, Japan). Seventy-two hours after MCAO, mice were euthanized with an overdose of isoflurane. Brains were immediately collected, frozen in 2-methyl butane chilled on dry ice, and stored at −80 °C until further histological analysis. The brains were sectioned into 40-µm thick coronal sections with a cryostat. Sections from 8 levels with 1-mm intervals were mounted onto glass slides and stained with cresyl violet. Images of the brain sections were acquired using a Zeiss microscope system and quantified by the evaluators who were masked to the animal identities by using image analysis software MCID Basic 7.0 (InterFocus Imaging Ltd., Cambridge, UK). The infarct area and the total area of both contralateral and ipsilateral hemispheres were traced manually in the digital images and measured automatically by the computer. Direct infarct volume was determined by the sum of the ipsilateral infarct area divided by the total contralateral hemisphere volume, and infarct volume was expressed as a percentage of the contralateral hemispheric volume (Bay et al., 2018).
Stereotaxic LPS Injections
To elicit an inflammatory response, stereotaxic injections of 50 ng of LPS (Wang and Namura, 2011) were given bilaterally to C57BL/6 mice under control 12:12 LD schedules and after exposure to either four 6-h weekly advances or delays (n = 24 per group). One-half of each group (n = 12) received bilateral injections 1 week after the last lighting manipulation, between 0900 h and 1200 h (ZT 2–ZT 5), and the other half (n = 12) served as noninjected controls. LPS (Escherichia coli serotype 055: B5; Sigma-Aldrich, St. Louis, MO) was dissolved in sterile saline at a concentration of 50 ng/µL as described in Wang and Namura (2011). Mice were anesthetized with isoflurane 3% to 5% for induction and maintained at approximately 1% to 3%. Mice were placed into a rodent stereotaxic frame (David Kopf Instruments, Tujunga, CA) and burr holes were drilled 14 mm anterior to bregma, ±2.0 mm lateral to the midline, and 3 mm ventral to the surface of the skull. One microliter LPS (50 ng/µL) was injected bilaterally through a Hamilton micro syringe into the dorsal striatum over a 5-min period to avoid reflux along the injection track as used in past studies (Becchi et al., 2017). Mice woke up after the surgery and were sacrificed 6 h after injections to collect the brain tissue. RNA was promptly extracted from 1 hemisphere of the brain as previously described (Stowie et al., 2019) using Trizol/cholorform/ethanol extraction.
Quantitative Real-time Polymerase Chain Reaction
Gene expression analysis was done via quantitative polymerase chain reaction (qPCR) on a BIORAD CFX-96 real-time system using SYBR Green qPCR master mix from SABiosciences (Frederick, MD; Prod. No. PA-010). Proinflammatory markers used in this study were interleukin-1β (IL-1β; primer, F: CAACCAACAAGTGATATT, R: GATCCACACACTCTCCAGCT), IL-6 (primer, F: TAGTCCTTCCTACCCCAATTTCC, R: TGGTCCT-TAGCCACTCCTTC), and tumor necrosis factor–α (TNF–α; primer, F: ACGGCATGGATCTCAAAA, R: TGGGAGTAGACAAGGTAC). The anti-inflammatory marker used in this study was IL-10 (primer, F: GGGCCAGTACAGCCGGGA, R: CACCTGGCTGAAGGCAGT). Genes were amplified and reported as relative expression of transcript level and normalized to beta-2-microtubulin (primer, F: TGGTGCTTGTCTCACTGA, R: TTCAGTATGTTCGGCTTC) (Piehler et al., 2010).
Statistical Analysis
Statistical analysis of infarct volume was performed by Kruskal-Wallis analysis of variance (ANOVA) followed by Dunn’s post hoc analysis. Body temperature and rCBF were analyzed by 1-way analysis of variance. Gene expression analysis was quantified by the comparative 2−ΔΔCt method and is reported as relative to noninjected mice kept in controlled lighting conditions. Gene expression results were compared among groups by 1-way ANOVA and Tukey’s post hoc test. Cytokine ratios were analyzed using Kruskal-Wallis ANOVA and Dunn’s post hoc analysis. In all cases, p < 0.05 was considered statistically significant.
Results
Stroke Severity Is Affected by ECD
There were no significant differences in body temperature or CBF among the groups (Table 2), suggesting that all mice had similarly successful MCAO surgeries. Five of 65 animals were excluded from infarct volume measurement: 3 were euthanized before completing the study because of poor general condition that did not respond to supportive treatments; in 1 animal, the filament could not be retrieved from the carotid artery; in another, reperfusion was not sufficient. Statistical analysis suggests an overall group difference in direct infarct size (p = 0.011), with post hoc analysis revealing an effect for group B (Dunn’s post hoc analysis, p < 0.05) with a near doubling of median infarct volume versus control (group A; Fig. 1). The infarct volume was not different from controls (group A) when the period following the phase advance schedule was extended from 1 to 3 weeks (group C), after a single phase advance (group D), or after the 4 weekly phase delays (group E).
Body temperature and regional cerebral blood flow (rCBF).
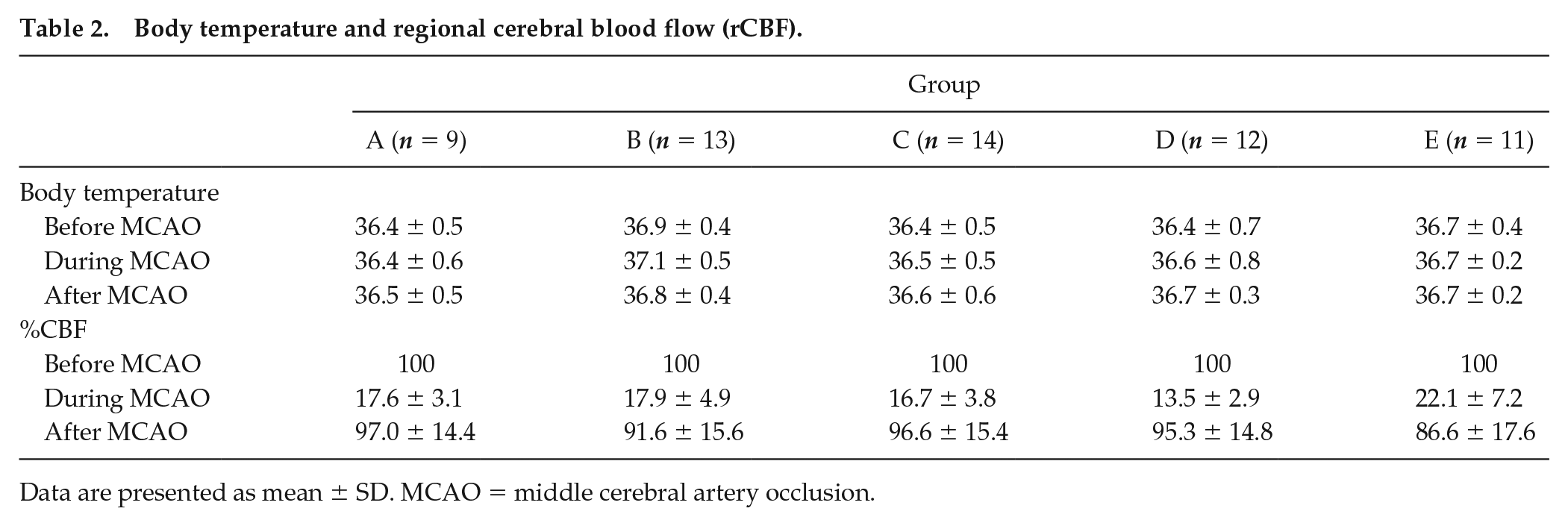
Data are presented as mean ± SD. MCAO = middle cerebral artery occlusion.
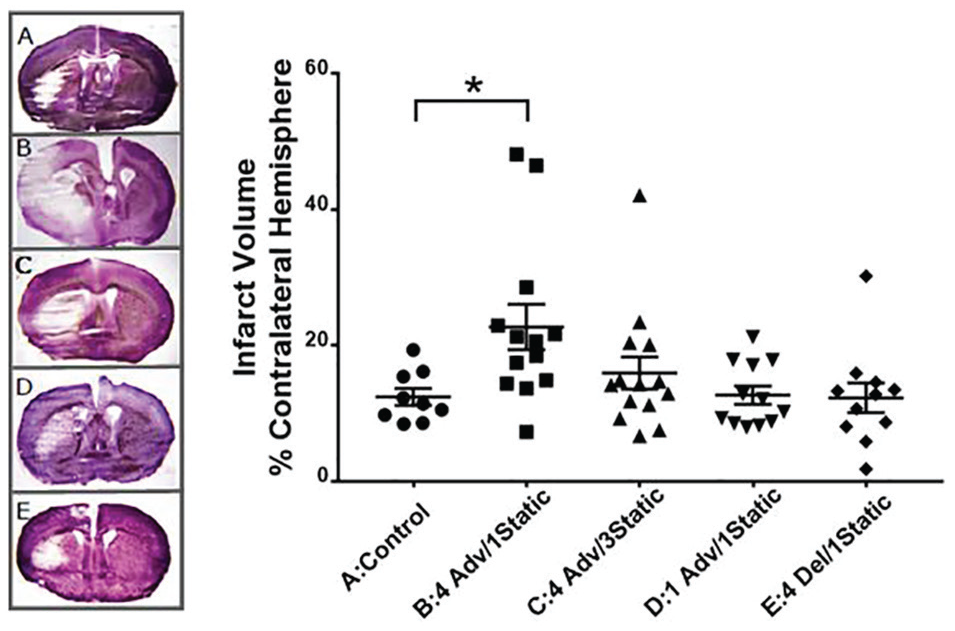
Effect of environmental circadian disruption schedules on cerebral infarction after transient focal ischemia with middle cerebral artery occlusion in mice. Infarct volume of each ipsilateral hemisphere as a percentage of the contralateral hemisphere in cresyl violet–stained coronal brain slices. (A) Control (constant 12:12 light cycle; n = 9); (B) 4 week advance, 1 week static (n = 13); (C) 4 week advance, 3 week static (n = 14); (D) 1 week advance, 1 week static (n = 12); (E) 4 week delayed, 1 week static (n = 11). One representative example from each group is also shown. Values are significantly different (Kruskal-Wallis nonparametric analysis of variance, p = 0.011; Dunn’s post hoc analysis, p < 0.05) between the control group (A) and 4 weeks advanced, 1 week static group (B).
ECD Causes a Shift-dependent Increase in the Neuroinflammatory Response
Inflammatory cytokine expression was measured in brains taken from mice exposed to 4-weekly phase advances, 4-weekly phase delays, and control 12:12 lighting. Mice were sacrificed either 6 h after LPS injection or during the same time point without injection.
Noninjected mice were compared across groups for cytokine transcript levels (Fig. 2a). Levene’s test showed equal variance, and 1-way ANOVA was used to compare all groups for pro- and anti-inflammatory gene expression followed by Dunnett’s post hoc comparisons against controls. Whereas IL-6 and IL-1β were not significantly different across the groups (IL-6: F2,21 = 0.4527, p = 0.6420; IL-1β: F2,21 = 0.1805, p = 0.8361), mice in the delayed group showed a significant increase in IL-10 (F2,21 = 9.637, p = 0.0011). Multiple comparisons showed that the advanced group (mean = 0.4064, SEM = 0.9102) was not significantly higher than in the control group (mean = 1.824, SEM = 0.9102). However, the phase-delay group (mean = 4.351, SEM = 0.9102) did differ significantly from the control. TNF-α was also significantly increased after phase delays at baseline (F2,21 = 14.02, p = 0.0001); transcript levels with multiple comparisons showed that the advanced group (mean =16.96, SEM = 15.07) did not differ from control, but delays (mean = 88.28, SEM = 86.83) did.
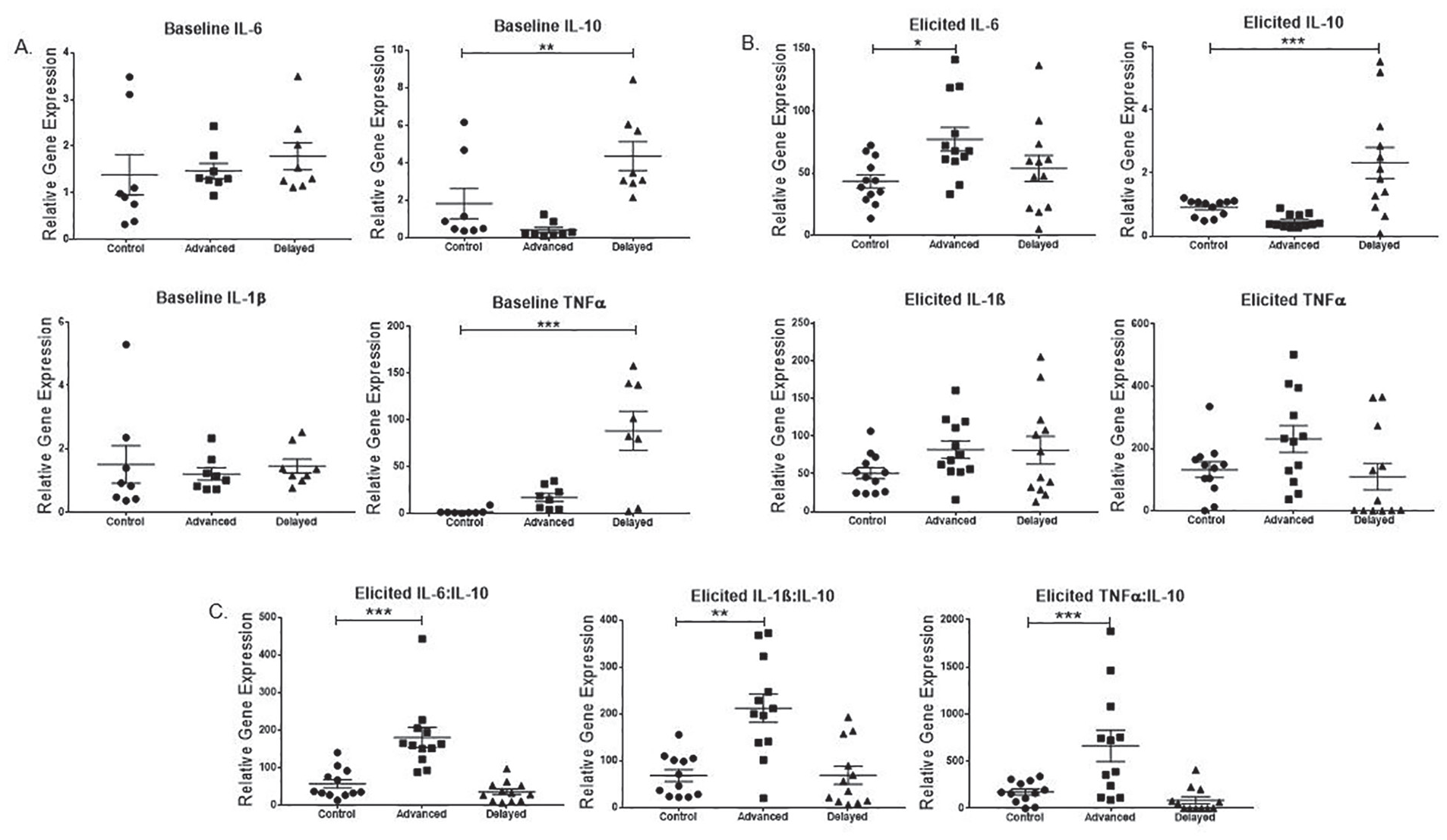
History of phase-advancing light conditions exacerbates lipopolysaccharide (LPS)–induced inflammatory cytokine transcript level. Male mice received either no injection (n = 8/gp) or 50 ng/mL LPS stereotaxic brain injections (n = 12/gp) after a history of exposure to either control (constant 12:12 light cycle), advanced (4 weekly 6-h advanced), or delayed (4 weekly 6-h delayed) light cycles. (A) Mean and individual baseline mRNA transcript levels were compared across noninjected mice exposed to all 3 conditions. Interleukin-6 (IL-6) and IL-1β did not differ among groups at baseline, but both tumor necrosis factor-α (TNF-α) and IL-10 were higher in the delayed group than in the unshifted controls. (B) After elicitation by LPS injection, the IL-6 transcript level increased significantly in the phase-advanced group compared with controls. IL-10 was significantly increased in delayed versus controls. IL-1β and TNF-α did not differ among groups. (C) Ratios of pro- to anti-inflammatory cytokine transcript levels showed phase-advanced mice had a dysregulated response in all 3 comparisons: IL-6:IL-10, IL-1β:IL-10, and TNF-α:IL-10. p values reflect post hoc comparisons against the relevant control group: *p ≤ 0.05; **p ≤ 0.001; ***p ≤ 0.0001.
Following elicitation by LPS injection, IL-6 transcript was highest in the phase-advanced group among the 3 conditions (F2, 33 = 3.934, p = 0.029). Post hoc multiple comparisons indicated that the advanced group (M = 77.207, SEM = 9.494) was significantly higher than the control group (M = 43.487, SEM = 5.251). However, the phase-delay group (M = 53.796, SEM = 10.487) did not significantly differ from the control conditions. IL-10 gene expression was significantly increased in the phase-delayed compared with the phase-advanced and control groups. One-way ANOVA (p < 0.001), followed by Tukey multiple comparison, indicated a significant difference between both control versus advanced and advanced versus delayed groups (p < 0.05). Both IL-1β and TNF-α showed no significant difference among the three experimental groups. A Kruskal-Wallis with Dunn’s post hoc analysis comparing the ratios of pro- to anti-inflammatory cytokine expression showed a dysregulated response in the phase-advanced group versus controls and delay groups (Fig. 2C). Ratios of both IL-6 to IL-10 (H = 22.01, p < 0.0001) advanced versus control (p = 0.001), and IL-1β to IL-10 (H = 13.88, p = 0.001) advanced versus control (p = 0.0065) differed significantly. The ratio of TNF-α to IL-10 response was also significantly different (H = 14.53, p < 0.0007); however, Dunn’s post hoc analysis showed only a trend toward dysregulation in controls versus advanced conditions (p = 0.0571).
Discussion
Shift work has been shown to be associated with wide-ranging deleterious health effects, including cerebrovascular diseases such as stroke. Shift workers have schedules that result in ECD, which has been associated with an increased risk of ischemic stroke that correlates to length of career (Brown et al., 2009). While causal relationships are difficult to establish in human studies, animal models can provide further insight. The functions of organ systems are regulated by circadian rhythmicity, and these daily changes are important for health and resilience. Altered light cycles cause a disruption of circadian rhythms, leading to a misalignment of peripheral clocks with negative consequences on healthy organ function (West et al., 2017a). In fact, advancing light schedules alone were sufficient to increase mortality in aged mice (Davidson et al., 2006). Here, we show that disrupting circadian rhythms via four 6-h weekly phase advances of the 12:12 light cycle, mimicking weekly eastward travel across 6 time zones (and backward rotating shift work), almost doubled the infarct volume compared with controls following surgically induced ischemic stroke in mice. Increasing the time following the phase-advanced lighting schedule to 3 weeks was enough to ameliorate the increased infarct volume after four 6-h weekly phase-advance schedules. Hence, the effects are not permanent. A single-phase advance was insufficient to produce the worsened outcome, suggesting that a more chronic exposure is necessary to induce changes. This observation is consistent with our earlier work, in which 4 weekly advances, but not 1 advance, induced changes in inflammatory signaling following peripheral LPS injection (Castanon-Cervantes et al., 2010). Furthermore, four 6-h weekly phase delays, mimicking forward rotation of shift schedules (i.e., westward travel), did not worsen stroke outcome. This finding is also consistent with our previous studies indicating that advancing schedules lead to worse outcomes than delaying schedules (Davidson et al., 2006; Castanon-Cervantes et al., 2010). Thus, specific work or travel schedules appear to be more harmful than others, and chronic exposure seems to be worse than a single experience of ECD. The mechanisms for why different light shifting schedules result in different outcomes are still unknown but may be related to the longer duration of misalignment and behavioral transients during advance shifting compared with delaying (Monk et al., 2000; Aschoff et al., 1975).
In this study, we did not quantify behavioral rhythmicity with running wheels since exercise can modify the effects of the circadian disruption manipulation (unpublished observations). We have, however, used these exact schedules as our primary method of ECD in previous publications where phase measures were used. We have shown that a 6-h phase advance results in temporary misalignment among central and peripheral oscillators, as well as misalignment within the central pacemaker itself during resynchronization (Sellix et al., 2012). However, the 7-day interval we use between shifts of the LD cycle is sufficient to allow for reentrainment of locomotor activity, sleep/wake rhythms, and peripheral oscillator phase (Castanon-Cervantes et al., 2010). This is an important factor because the stroke insult and others we have used have known phase-dependent effects, and our approach ensures that mice are fully reentrained prior to stroke. This schedule results in moderate sleep loss that does not seem to, alone, drive the immunomodulatory effects of ECD (Brager et al., 2013). The advancing schedule does not seem to induce stress hormones (Davidson et al., 2006) or behavioral correlates of anxiety or depression (Castanon-Cervantes et al., 2010). After such a 6-h advance, the rhythm in body temperature remains but runs a bit faster over 5 to 6 days to return to a normal phase angle of entrainment (Castanon-Cervantes et al., 2010). Unlike more severe/rapid schedules used in other laboratories (Halberg, 1975; Penev et al., 1998; Filipski et al., 2004; Preuss et al., 2008), rhythms are not abolished or significantly perturbed, and mice seem to shift in a similar manner to one another. We are, perhaps, just forcing the clock to run a bit faster or a bit slower, rather than causing a profound desynchrony as would be the case in a Bmal1-/- mouse, for example (Stowie et al., 2019)., Thus describing the protocol as inducing circadian disruption is perhaps an oversimplification, as the term more refers to the stimulus (i.e., repeated shifting of the LD cycle) than of its consequences. As a method to isolate the specifically circadian consequences of shift work in an inbred mouse strain, this model has value.
Our study builds on a growing body of evidence that stroke risk/severity is linked to circadian disruption. Earnest and colleagues (2016) showed that phase advancing light cycles increased stroke severity in rats after MCAO surgery using endothelin-1 to induce prolonged ischemia with gradual reperfusion. In contrast, a recent study determined that there was no increase in infarct size after a permanent MCAO using diathermy after circadian disruption (Ku Mohd Noor et al., 2017). Such different outcomes may be due to the different circadian disruption schedules used, as we have shown the schedule dependence of such effects. Different outcomes may also have arisen because of the relatively mild insult we employed: a temporary (30-min) occlusion immediately followed by reperfusion, allowing for an increase in infarct to occur in some groups versus others. The permanent occlusion model used by Ku Mohd Noor and colleagues (2017) may have resulted in a ceiling effect.
The circadian clock regulates immune function. The circadian clock gene Rev-erbα has been shown to mediate inflammatory responses of proinflammatory cytokines in macrophages (Gibbs et al., 2012). The expression of chemokine CCL2 is regulated by Rev-erbα gene expression and exhibits increased expression in rev-erbα knockout mice. Chronic and systemic inflammation dampened Rev-erbα gene expression in peritoneal macrophages and suppressed CCL2-activated signals in the inflammation signaling cascade by impairing cell adhesion and migration (Sato et al., 2014). Circadian disruption elevated proinflammatory IL-17–producing CD4+ T-helper cell expression in the intestine, increasing susceptibility to inflammatory disease (Yu et al., 2013). Uncontrolled or chronic inflammation is a risk factor shared among the pathologies observed in both shift workers and animal models of ECD. Specifically, inflammation following stroke has been shown to increase damage and neurological deficit (Chapman et al., 2009). The acute local inflammatory responses to stroke are characteristic of brain ischemia and mediated by cytokines (Vila et al., 2003). Neurodegeneration because of poststroke inflammation results in the expansion of the ischemic lesion (Simats et al., 2016). Successful time of delivery of drugs that induce reperfusion after acute ischemic stroke has been shown to decrease the debilitating effects of stroke damage (Chevilley et al., 2015). Therefore, the increased infarct volume may present a notable treatment challenge in acute stroke patients with a history of shift work. The proinflammatory cytokine IL-6 is elevated by ECD in a schedule-dependent manner in mice in vivo (Castanon-Cervantes et al., 2010), in peripheral immune cells harvested from ECD-exposed mice (Adams et al., 2013), and in spleen exposed to ECD in vitro (Stowie et al., 2019). In this present work, our results suggest that a chronically advancing light schedule leads to heightened IL-6 signaling at baseline and in response to an innate immune stimulus provided directly to the brain. Phase-delaying schedules did not increase IL-6 responses but instead caused a heightened anti-inflammatory (IL-10) response before and after LPS challenge. The change in IL-10 expression during baseline (before LPS injection) suggests that the delay group seems to be primed to quickly respond to/inhibit inflammatory signals and may explain why this group had a smaller infarct size. IL-10 directly downregulates IL-6 and TNF-α, but its effects are not confined to these intermediaries (Stenvinkel et al., 2005). One might speculate that IL-10 is responsible for a “protected” response in this context, but the complexity and interplay among innate immune signaling molecules and cells is complex and context specific, making firm conclusions difficult to draw. For example, TNF-α is a master regulator of the cytokine cascade critical to mounting a swift defense against infection but can be fatal when excessively upregulated (Stenvinkel et al., 2005). Regulation of IL-10 expression is even more complex than that of TNF-α (Meisel et al., 1996). Interestingly, these baseline changes in cytokine expression after ECD were not observed in our previous studies. When pro- and anti-inflammatory mediators are considered together using a ratio approach relevant to disease outcome (Biswas et al., 2010), it becomes clear that advancing ECD primes the innate immune system in the brain towards an M1-type response that likely exacerbates the cell death associated with focal ischemia (Frank-Cannon et al., 2009). Therefore, we suspect that the increased severity of ischemic damage may be caused by altered innate immune function and increased inflammation, often referred to as priming, by previous exposure to ECD. Such a conclusion would be further supported by data we do not provide: a demonstrated increase in inflammatory cellular markers after ECD and LPS treatment and measurement of cytokine protein levels to further validate our mRNA measurements. In addition, if priming were responsible for the imbalanced response and increased infarct, we would expect that such cytokine effects would be absent after a single phase advance and would also disappear in the longer recovery group. These 2 groups, for which infarct volume was similar to controls, were not included in the cytokine experiment.
In conclusion, we show that ECD leads to worsened stroke outcome, likely because of a disrupted balance between pro- and anti-inflammatory cytokine gene expression. This observation is consistent with a compelling body of evidence that disruption of circadian rhythmicity by exogenous environmental factors affects disease resilience (Evans and Davidson, 2013). Our evidence suggests that the health impacts of shift work may be improved by targeting shift work scheduling, inflammatory pathways, or a combination of efforts. Ultimately, these findings may prove to be important for the health management and scheduling considerations for people with lifestyles that include chronic disturbances of circadian rhythmicity such as frequent jet lag and shift work.
Footnotes
Acknowledgements
We acknowledge Fang Du, MD, for administration of surgical MCAO and Qingmin Guo, PhD, Donghui Li, PhD, and Shobu Namura, MD, for direction in study design and analysis. We also thank Dr. Kenkichi Baba and Sydney Pitts for technical assistance, Chaohua Li for advice on statistics, Morehouse School of Medicine Neuroscience Institute, and Morehouse School of Medicine RCMI G12 program for their support. This work was supported by National Institutes of Health (NIH) G12MD007602 to C. Bond, NIH SC1GM112567 to A. Davidson, NIH U54NS083932 to P. MacLeish, NIH SC2GM125493 to O. Castanon-Cervantes, and NIH R25GM058268 to S. Harris-Hooker.
Conflict of Interest Statement
The authors have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.