
Abstract
Casein kinase 1ε (CK1ε) performs key phosphorylation reactions in the circadian clock mechanism that determine period. We show that the central clock protein PERIOD2 (PER2) not only acts as a transcriptional repressor but also inhibits the autoinactivation of CK1ε, thereby promoting CK1ε activity. Moreover, PER2 reciprocally regulates CK1ε’s ability to phosphorylate other substrates. On output pathway substrates (e.g., P53), PER2 inhibits the activity of CK1ε. However, in the case of central clock proteins (e.g., CRYPTOCHROME2), PER2 stimulates the CK1ε-mediated phosphorylation of CRY2. CK1ε activity is temperature compensated on the core clock substrate CRY2 but not on output substrates, for example, the physiological output protein substrate P53 and its nonphysiological correlate, bovine serum albumin (BSA). These results indicate heretofore unrecognized pivotal roles of PER2; it not only regulates the central transcription/translation feedback loop but also differentially controls kinase activity CK1ε in its phosphorylation of central clock (e.g., CRY2) versus output (e.g., P53) substrates.
The circadian biological clock regulates myriad intracellular processes that include cell division and proliferation (Matsuo et al., 2003; Johnson, 2010). Moreover, investigations of DNA damage responses have revealed that nucleotide excision repair, DNA damage checkpoints, and apoptosis are appreciably influenced by this biological clock (Sancar et al., 2015). An ensemble of different molecular components participates in a feedback loop of clock proteins that are rhythmically abundant and interact with each other to modulate their activities. The current model for the molecular mechanism of the mammalian clockwork proposes autoregulatory transcriptional and translational feedback loops (TTFLs) of central clock gene expression (Hardin et al., 1990; Reppert and Weaver, 2002). Rhythmic transcriptional “drive” is provided by the positive bHLH-PAS transcription factor BMAL1, which dimerizes with CLOCK (Hogenesch et al., 1998). These BMAL1/CLOCK heterodimers activate transcription primarily at E-boxes (Gekakis et al., 1998). Negative feedback has been thought to be accomplished by Period and Cryptochrome gene products (PER1/2/3 and CRY1/2) (Reppert and Weaver, 2002). However, a recent publication reevaluates the relative role of PERs versus CRYs in directly repressing transcription at E-boxes; in this view, CRY is the dominant repressor in the TTFL, and PER causes the dissociation of the BMAL1/CLOCK complex from E-boxes by interacting with CRY in a process that depends on the CKBD (casein kinase binding domain) of PER (Ye et al., 2014).
Casein kinases 1δ and 1ε (CK1δ/ε) are essential components of the mammalian clockwork that significantly determine circadian period and reentrainment kinetics (Eide and Virshup, 2001; Eide et al., 2002; Lee et al., 2011; Pilorz et al., 2014). Rhythmic posttranslational modifications—especially phosphorylation—are a pervasive feature of key clock proteins, but their role in the mammalian mechanism is ill-defined (Lee et al., 2001). The identification of CK1δ/ε as a key kinase phosphorylating central clock proteins came by the discovery that the tau mutant of hamster circadian rhythms was a mutation in the CK1ε gene (Ralph and Menaker, 1988; Lowrey et al., 2000). CK1δ/ε is homologous to Doubletime, which had already been implicated in key phosphorylation events in the Drosophila circadian clock mechanism (Kloss et al., 1998). CK1δ/ε and their homologs are now recognized to be evolutionarily conserved as key players in circadian and circatidal rhythmicity (Eide and Virshup, 2001; van Ooijen et al., 2013; Zhang et al., 2013), including having polymorphisms that are responsible for sleep phase syndromes in humans (Xu et al., 2005). CK1δ/ε is present in both nucleus and cytoplasm, but it is thought to associate with PER and CRY in the cytoplasm and facilitate PER/CRY translocation to the nucleus (Lee et al., 2001). Moreover, CK1δ/ε phosphorylates PERs, CRYs, and BMAL1, and this process may target those proteins for ubiquitination and degradation or, alternatively, may regulate their activity in clock-mediated transcription on E-boxes (Eide et al., 2005; Ye et al., 2014).
We report here that circadian control of gene expression is balanced with circadian phosphorylation mediated by CK1ε activity by pivotal activity of the clock protein PER2. PER2 inhibits the autophosphorylation of CK1ε and therefore promotes CK1ε activity. By virtue of its promiscuous phosphorylation of many substrate proteins, CK1ε regulates many circadian outputs, including cell division and proliferation. PER2 acts as an inhibitor of CK1ε-mediated phosphorylation of output substrates, including the nodal regulator P53. At the same time, PER2 stimulates the phosphorylation of clock protein substrates such as CRY. Because PER undergoes phase-dependent translocation to the nucleus, (PER) concentration changes dramatically over the circadian cycle in the nucleus versus the cytoplasm. Consequently, PER2 can affect CK1ε activity differentially over the cycle by concentration-dependent enzymatic effects. Finally, CK1ε activity is temperature compensated on full-length clock protein substrates (but not on the non–clock protein substrates P53, casein, and bovine serum albumin [BSA]), and therefore CK1ε activity can form a temperature-compensated timer of a portion of the circadian cycle.
Materials and Methods
Protein Expression and Purification
We used pGEX-6P-1 (Amersham Biosciences, Piscataway, NJ) to clone and express in Escherichia coli cells mPer2, mCry2, and CK1εΔC (Suppl. Figures S1-S6; primers used are listed in Suppl. Table S1). Other protein expression plasmids were pGEX-human p53 (ID 24860; Addgene Cambridge, MA), PP1 (ID 26566; Addgene), and pRSET-CK1ε. Enzymes used for cloning are from New England Labs (BioLabs, Ipswich, MA). The mPER2 and mCRY2 proteins were expressed in E. coli strain Rosetta2 cells (71402; Novagen Danvers, MA) and purified as described in Supplemental Figures S2 to S5. The GST fusion CK1εΔC proteins were expressed in E. coli strain BL21 cells and purified with glutathione agarose beads (16100; Pierce Rockford, IL) followed by cleavage with PreScission Protease (27-0843-01; GE Healthcare, Piscataway, NJ). The eluted CK1εΔC proteins were further purified by cation exchange purification with SP Sepharose beads (17-0657-03; GE Healthcare). The GST-hP53 and hPP1 proteins were purified as described (Ayed et al., 2001; Kelker et al., 2009), and the CK1ε-FL proteins were purified as previously reported (Eide et al., 2005). The concentration of each protein was measured with the Bradford reagent (Bio-Rad Protein Assay; Bio-Rad, Hercules, CA) using a dilution series of BSA (Bio-Rad) to generate a standard curve. The purity of each protein was determined on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Suppl. Figure S1).
CK1ε Kinase Phosphorylation Assays
To assess inhibition/enhancement effects of mPER2 on CK1ε activity, 20 µL of 50 nM CK1ε-ΔC or CK1ε-FL (1 µM CK1ε-FL was dephosphorylated by 0.01 µM hPP1 one day before the experiment) was incubated with different substrates (0.5 µM or 5 µM BSA, 1 µM mCRY2, or 1 µM GST-hP53) in Kinase Reaction Buffer (20 mM Tris-Cl, 10 mM MgCl2, 5 mM dithiothreitol [pH 8.0], diluted from a 5× master mix), in the presence of increasing concentrations of mPER2 proteins (range, 0-300 nM). In total, 1 mM ATP (10519979001; Roche, Basel, Switzerland) and 0.1 µCi/µL ATP-γ-32P (NEG002A250UC; PerkinElmer, Waltham, MA) were included in the reaction buffer. Reactions were performed at 30 °C for 3 h and stopped by adding 5 µL of 5× SDS loading buffer. mPER2 inhibitory effects on the kinase activity of CK1ε-FL were tested by a 1-h time course incubation of 20 nM CK1ε-FL with either 1 µM GST-hP53 or 300 nM mPER2. mPER2 effects on the kinase activity of CK1ε were tested by incubation of various substrates (BSA, mCRY2, or GST-hP53) with 50 nM CK1ε-ΔC in the absence or presence of 50 nM mPER2 (100 nM mPER2 was used when mCRY2 was the substrate). Temperature effects on CK1ε-ΔC activity were tested at 25 °C and 35 °C with the substrates GST-hP53 and mPER2 (20 nM CK1ε-ΔC was incubated with 1 µM GST-hP53 or 300 nM mPER2). The reaction products were resolved by 10% SDS-PAGE, and the resulting gels were stained with colloidal Coomassie brilliant blue (CBB). Autoradiography of the dried gels was assessed by a PhosphorImager (Amersham Biosciences).
Results
PER2 Has Reciprocal Feedback on CK1ε Activity
CK1ε has a C-terminal domain that it autophosphorylates, rapidly turning off its own activity (Graves and Roach, 1995; Cegielska et al., 1998; Rivers et al., 1998). Therefore, most of the assays reported for CK1ε use a protein in which the C-terminal autoinhibitory domain is removed, herein called CK1ε-ΔC, to distinguish it from the full-length CK1ε kinase protein, which will be named in this study “CK1ε-FL.” The mPER2 protein is a substrate of CK1ε-ΔC–mediated phosphorylation (Lowrey et al., 2000; Toh et al., 2001), but in addition, mPER2 stimulates CK1ε-ΔC–mediated phosphorylation of the central clock proteins BMAL1 and mCRY1/2 (Eide et al., 2002). This action of CK1ε on BMAL1 modulates the transcriptional activity of circadian-regulated E-box promoters (Eide et al., 2002). We discovered that mPER2 not only stimulates the CK1ε-mediated phosphorylation of the clock protein substrates BMAL1 and mCRY1/2 but also acts as an inhibitor to CK1ε-mediated phosphorylation of non–clock proteins. Traditionally, the milk protein casein was used as a substrate to assay the activity of casein kinases (hence their name), but we have used as non–clock protein substrates BSA and the physiological substrate P53 (Knippschild et al., 1997; Dumaz et al., 1999). Figure 1A-D shows that CK1ε-ΔC–mediated phosphorylation of BSA is progressively inhibited by increasing concentrations of mPER2 protein. Based on 2 different concentrations of BSA (0.5 and 5 µM), the IC50 for mPER2’s inhibition is in the range of 30 to 50 nM mPER2. The same experiment was performed with CK1ε-FL to confirm that the full-length protein responds similarly to CK1ε-ΔC, and again mPER2 was found to inhibit CK1ε-FL–mediated phosphorylation of BSA in a similar range of IC50 (Figure 1E,F). This inhibition is specific for PER2 because increasing concentrations of P53 do not inhibit CK1ε-mediated phosphorylation of BSA (Suppl. Figure S7). Moreover, when the physiological substrate hP53 is used as a substrate, similar results are obtained. PER2 inhibits phosphorylation of hP53 by CK1ε-ΔC (IC50 ~36 nM; Figure 2A,B) and CK1ε-FL (IC50 ~47 nM; Figure 2C,D).
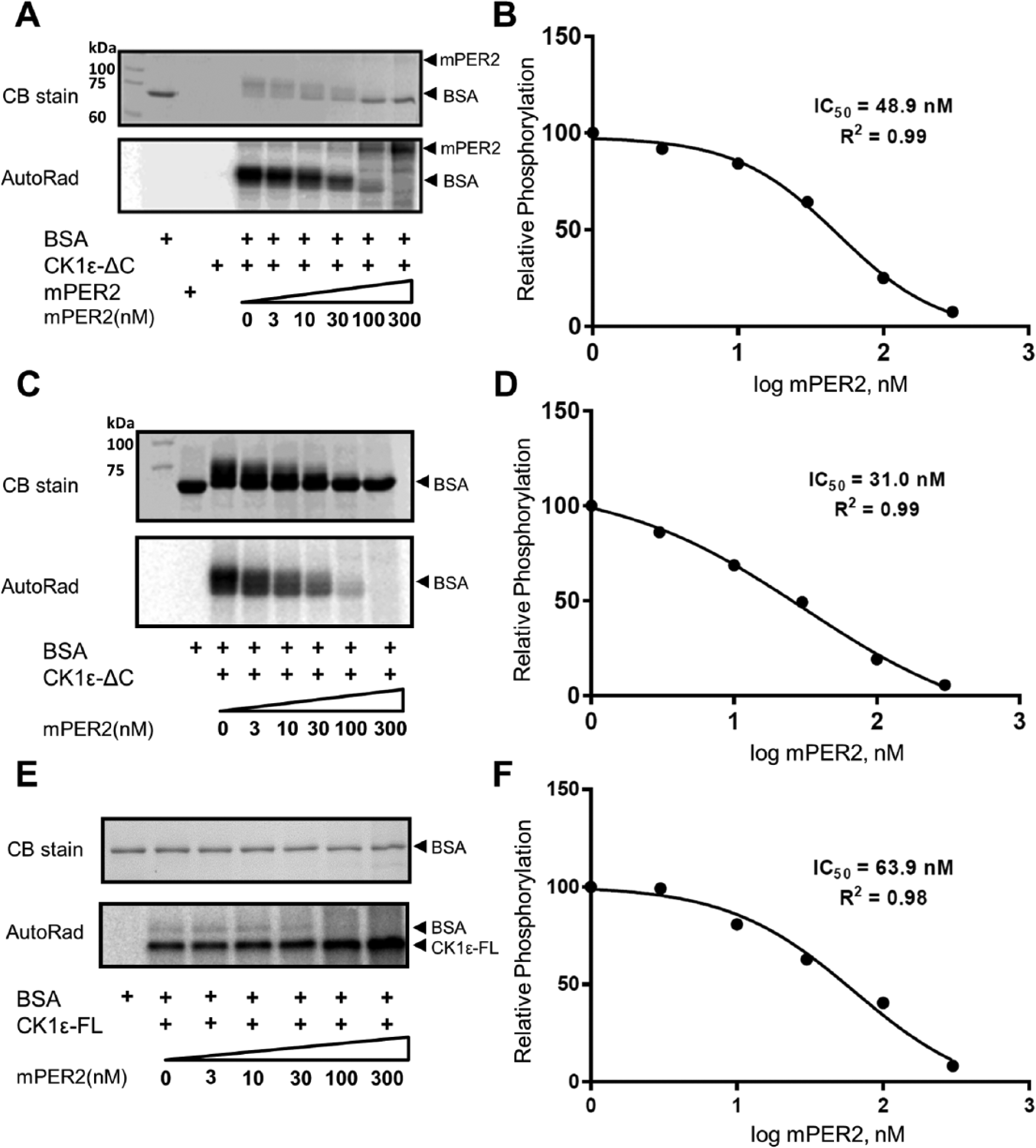
PER2 inhibits CK1ε activity in vitro. (A) Phosphorylation of bovine serum albumin (BSA) by CK1ε is inhibited by the addition of purified mPER2 in a dose-dependent manner. The phosphorylation of BSA (0.5 µM) is indicated by the mobility shift in the Coomassie blue (CB)–stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel or by incorporation of P-32 and autoradiography (AutoRad). mPER2 is also a substrate of CK1ε, as indicated by phosphorylation of the higher MW band in the AutoRad image. (B) Quantification of the mPER2 inhibition from panel A. The concentration of mPER2 that achieves 50% inhibition of BSA phosphorylation by the catalytic domain of CK1ε (CK1εΔC) is calculated as 48.9 nM. (C) Same as panel A, except with 10× concentration of BSA (5.0 µM). (D) Quantification of the mPER2 inhibition in panel C, where the IC50 is calculated as 31.0 nM (comparison of panels B with D indicates that mPER2 is not a competitive inhibitor). (E) mPER2 also inhibits full-length CK1ε (CK1ε-FL). hPP1-dephosphorylated CK1ε-FL was used as the kinase for this BSA phosphorylation assay. In addition, CK1ε autophosphorylates itself (see AutoRad). (F) Quantification of the mPER2 inhibition of full-length CK1ε in panel E; IC50 is calculated as 63.9 nM. (n = 2 for all panels; comparable results were obtained in both experiments.)
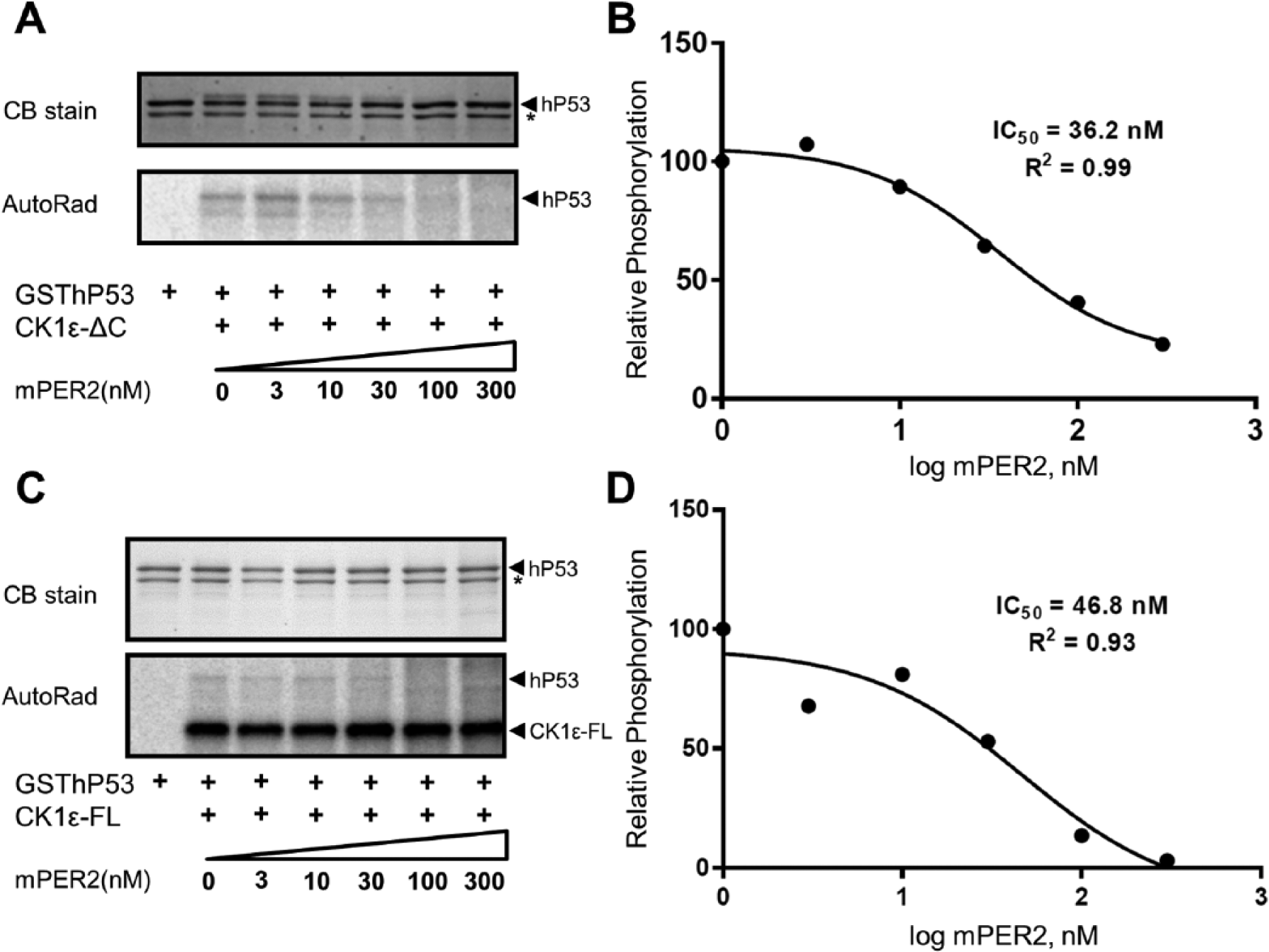
PER2 inhibits CK1ε-mediated phosphorylation of hP53. (A) hP53 is a substrate of CK1ε, and the phosphorylation of a hP53-GST fusion protein (GSThP53) is inhibited by the addition of purified mPER2. The phosphorylation of GSThP53 is indicated by the mobility shift in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels (Coomassie blue [CB] stain) or by P-32 incorporation (AutoRad). (B) Quantification of data depicted in panel A, where the IC50 of mPER2 to GSThP53 phosphorylation by CK1εΔC is calculated as 36.2 nM. (C) Phosphorylation of GSThP53 by CK1ε-FL is also inhibited by mPER2 (AutoRad), and CK1ε-FL autophosphorylates as in Figure 1. (D) Quantification of data depicted in panel C, where the IC50 for the GSThP53 substrate is calculated as 46.8 nM. (n = 2 for all panels; comparable results were obtained in both experiments.) The asterisk in the “CB stain” portions of panels A and C refers to a contaminating protein that co-purified with hP53.
PER2 Relieves Autoinhibition of CK1ε and Stimulates Its Phosphorylation of CRY2
Full-length CK1ε-FL autoinhibits itself within minutes by autophosphorylation (Graves and Roach, 1995; Cegielska et al., 1998; Rivers et al., 1998). Previous studies proposed that protein phosphatases such as PP1 maintain the activity of CK1ε-FL in vivo (Rivers et al., 1998; Cegielska et al., 1998). However, the data of Figure 1E,F and 2C,D are compatible with the possibility that PER2 also prolongs CK1ε-FL activity. Indeed, we found that PER2 inhibits the rate of CK1ε-FL autophosphorylation in vitro (Figure 3). When 1 µM P53 is used as a substrate, CK1ε-FL autophosphorylates relatively rapidly (Figure 3A,C), but when 300 nM PER2 is used as a substrate, the rate of CK1ε-FL autophosphorylation is much slower (Figure 3B,C).
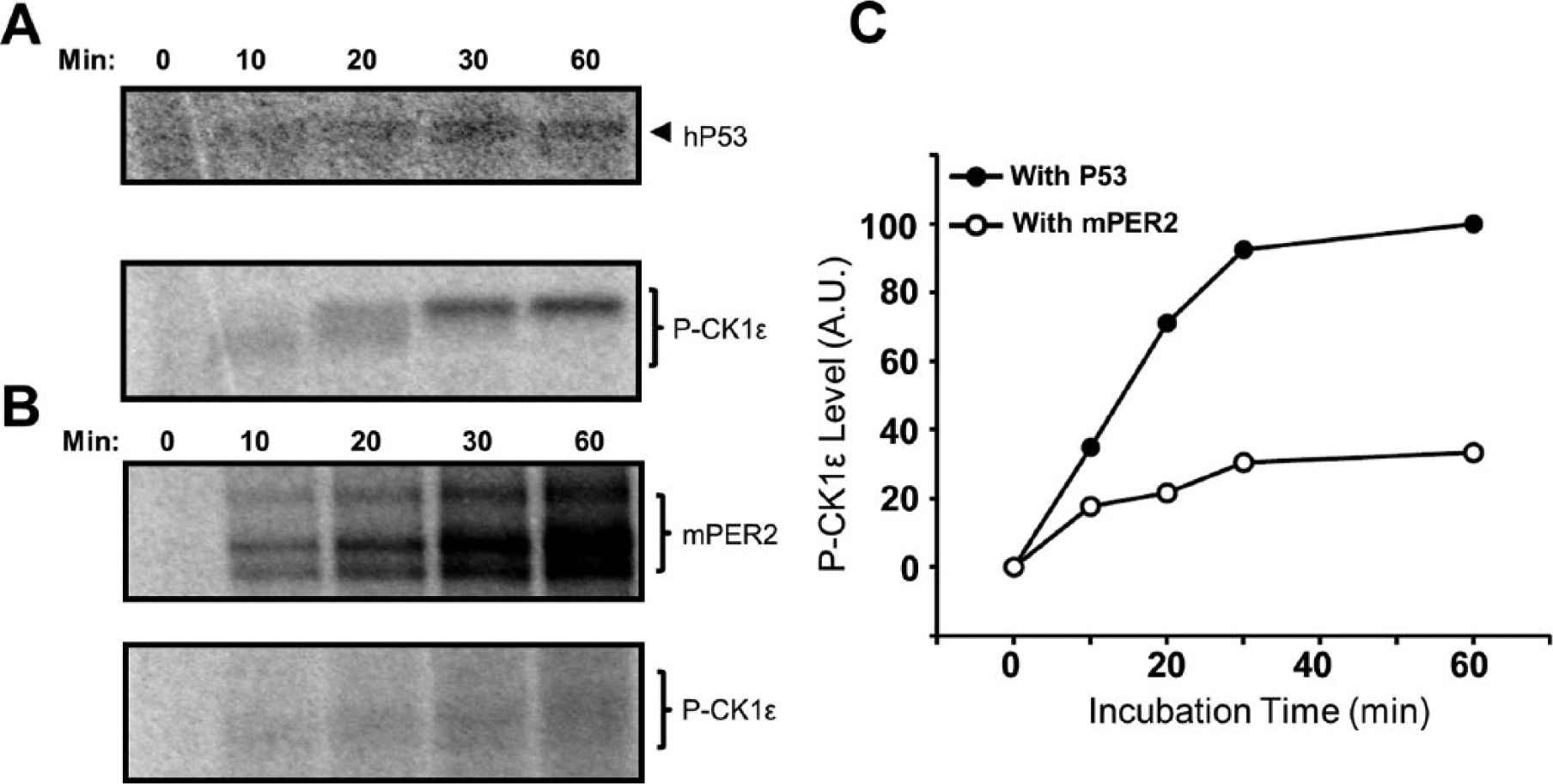
PER2 inhibits the rate of CK1ε-FL autophosphorylation. (A) CK1ε-FL rapidly autophosphorylates when it is incubated with 1 µM hP53, reaching its highest mobility shift within 30 min. hP53 is also phosphorylated by the full-length kinase CK1ε-FL, as indicated by the autoradiography image. (B) The autophosphorylation of CK1ε-FL is slower (in lower panel) when it is incubated with 300 nM mPER2. (C) Quantification of P-CK1ε levels from the autoradiography images in panels A and B.
Cell culture and cell lysate assays suggested that mPER2 enhances the interaction of the central clock protein CRY1 with CK1ε as well as stimulates CK1ε-mediated phosphorylation of CRY1 (Eide et al., 2002). We show in our purely in vitro assay that the CRY1 homolog, CRY2, is also phosphorylated by CK1ε and that CK1ε-ΔC–mediated phosphorylation of mCRY2 is increased when mPER2 is present in the assay (Figure 4A,C,D). In this assay, CK1ε-ΔC was used rather than CK1ε-FL because its high kinase activity yielded unambiguous results. This stimulation is concentration dependent, with an EC50 ~18 nM for mPER2 (Figure 4B). Therefore, mPER2 acts on CK1ε to reciprocally activate phosphorylation of central clock proteins (e.g., CRY2 in Figure 4) while inhibiting phosphorylation on non–clock protein substrates (e.g., BSA and P53; Figures 1-2).
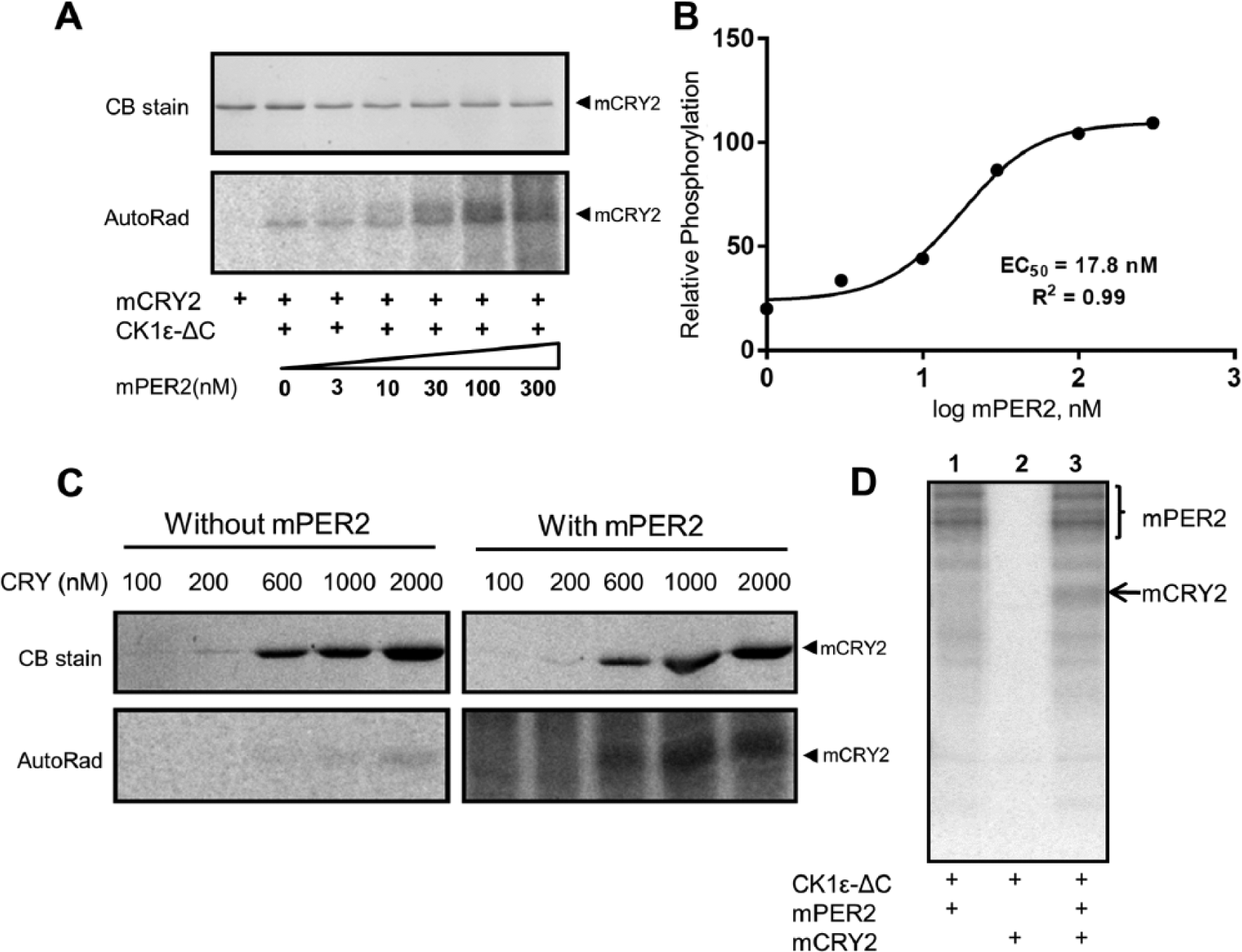
PER2 enhances CK1εΔC-mediated phosphorylation of CRY2. (A) CK1εΔC-mediated phosphorylation of mCRY2 is stimulated by the addition of purified mPER2 in a dose-dependent manner (AutoRad). (B) Quantification of the data depicted in panel A. The half-maximal effective concentration (EC50) for mPER2 stimulation of mCRY2 phosphorylation by CK1εΔC is calculated as 17.8 nM. (C) Phosphorylation of various concentrations of mCRY2 by CK1εΔC in the absence/presence of 100 nM mPER2. (D) mCRY2 is only phosphorylated by CK1ε in the presence of mPER2 (n = 2).
Substrate-Dependent Temperature Sensitivity/Compensation of CK1ε-ΔC
Because PER2 is known to be a phosphorylation substrate of CK1ε (Price et al., 1998; Lowrey et al., 2000; Toh et al., 2001; Shirogane et al., 2005), some authors have concluded that the period of the circadian oscillator is primarily determined by the phosphorylation status of PER2, which was proposed to be set by a balance between phosphorylation by CK1δ/ε and dephosphorylation by protein phosphatase 1 (Lee et al., 2011). Temperature compensation is a key defining property of circadian clocks (Q10 of most circadian rhythms ranges between 0.9 and 1.1), and it was pleasing that the rate of CK1ε-ΔC–mediated phosphorylation of a peptide substrate derived from PER2 (βTrCP-peptide) was reported to be nearly the same at 25 °C and 35 °C in vitro, whereas the same reaction is relatively temperature dependent for a non–clock protein substrate (casein) (Isojima et al., 2009). However, the significance of that report has been questioned since it tested substrates that were not physiological—namely, casein and a short peptide (derived from the PER2 sequence). We reexamined this important issue using the physiological substrate P53 and full-length PER2. CK1ε-ΔC–mediated phosphorylation of the non–clock protein P53 is relatively temperature dependent; compared at 25 °C versus 35 °C in vitro, the Q10 of the reaction is approximately 1.7 (Figure 5A,B).
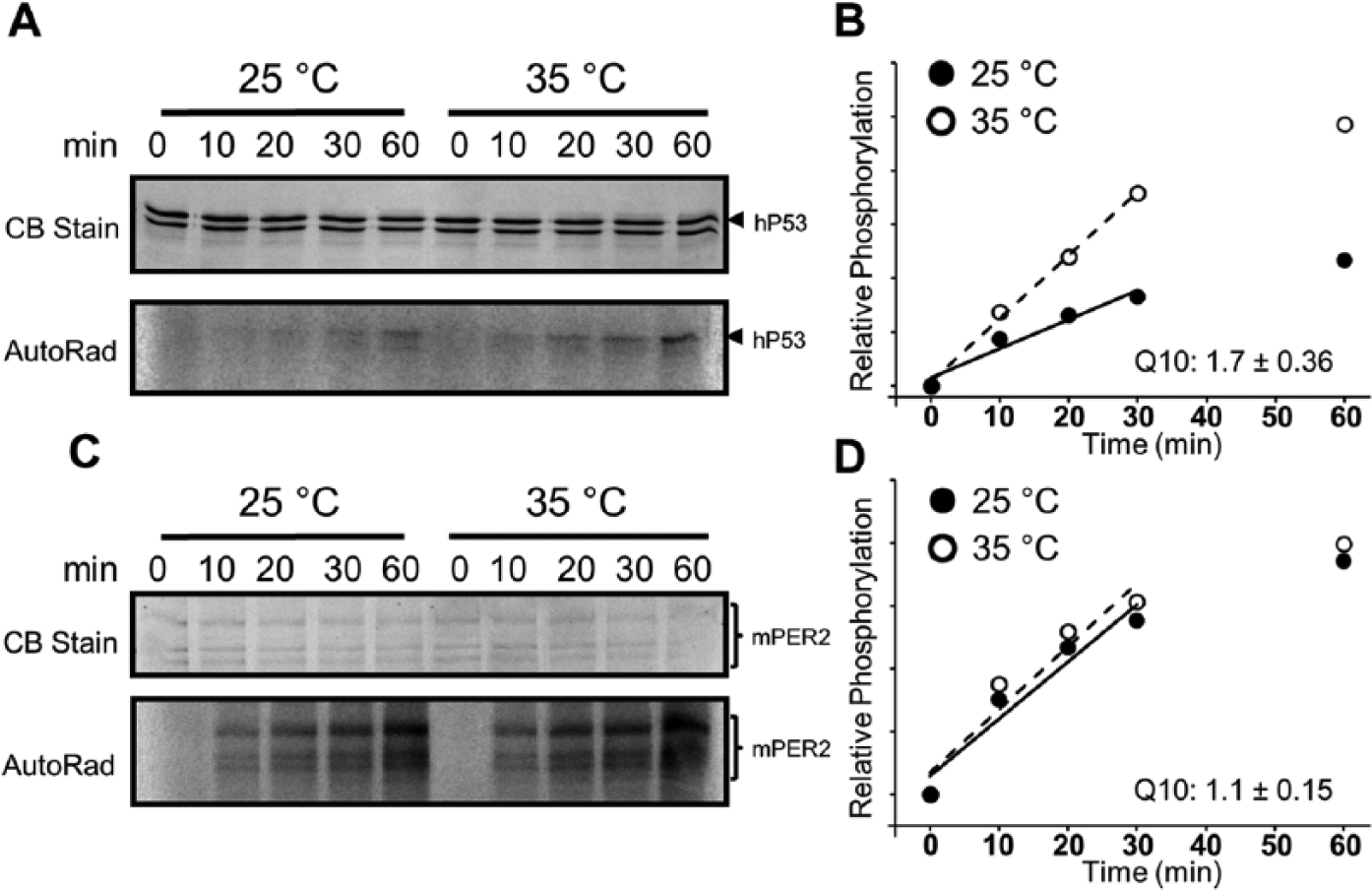
Substrate-dependent temperature compensation of CK1εΔC activity in the presence of mPER2. (A) Temperature dependency of CK1εΔC-mediated phosphorylation of the GSThP53 substrate, compared at 25 °C and 35 °C (AutoRad). (B) Quantification of CK1εΔC activity in panel A (closed circles = 25 °C and open circles = 35 °C). The data in the initial 30-min incubation were used to calculate the phosphorylation rates, and Q10 was calculated by dividing the rate at 35 °C over the rate at 25 °C. For the GSThP53 substrate, Q10 = 1.7 ± 0.36 (n = 4). (C) Temperature compensation for the CK1εΔC-mediated phosphorylation of the mPER2 substrate at 25 °C and 35 °C (AutoRad). (D) Quantification of CK1εΔC activity in panel C (closed circle = 25 °C and open circle = 35 °C). The data in the initial 30-min incubation were used to calculate the phosphorylation rates, and the Q10 for the mPER2 substrate was calculated as 1.1 ± 0.15 (n = 4). (See Suppl. Figure S8 for more information and an example of the replication of this experiment.)
On the other hand, when the full-length clock protein mPER2 is used as the substrate, the rate of CK1ε-ΔC–mediated phosphorylation is essentially temperature compensated between 25 °C and 35 °C (Q10 ~1.1; Figure 5C,D). This result extends the observations of Isojima and coworkers (2009) to demonstrate that the relative temperature independence of CK1ε-ΔC–mediated phosphorylation on peptide segments of PER2 is also true for phosphorylation on full-length PER2. Moreover, PER2 does not confer temperature compensation of CK1ε-ΔC–mediated phosphorylation on the non–clock substrates P53 and BSA; CK1ε-ΔC phosphorylates the BSA substrate with a Q10 ~3 (comparing 25 °C and 35 °C) whether PER2 is present or not (not shown). Therefore, it is the substrate interaction with CK1ε-ΔC that confers the temperature compensation, not the presence or absence of PER2.
Discussion
PER, CRY, and CK1δ/ε proteins are generally thought to associate in the cytoplasm, undergo phosphorylation reactions, and translocate to the nucleus (Lee et al., 2001). This study shows that PER modulates the activity of CK1ε, whereby it is an inhibitor on some key output substrates (e.g., P53) while at the same time stimulating the activity of central clock proteins that participate in the circadian TTFL. The action of PER2 on CK1ε obeys standard biochemical mass action enzymatic properties and is therefore strictly dependent on the concentration of PER protein. The concentration dependence of PER2 action is highly relevant to its possible role in regulating CK1ε-dependent clock outputs (such as cell division via P53) and the central TTFL because its concentration will undergo an approximately 10-fold increase as it translocates to the nucleus. While the absolute concentrations of PER in the cell are not known, the concentration of most transcriptional factors in the cell (cytoplasm + nucleus) lies in the range of 20 to 300 nM (see Suppl. Table S2), and PER concentrations are likely to lie on the low side of this range (Lee et al., 2001). Therefore, a 10× increase by translocation to the nucleus will bring the PER concentration into ranges similar to our in vitro experiments showing robust modulation of CK1ε activity (Figures 1 -4). Figure 6A depicts the concept of compartmentalized PER2 impact on CK1ε activity. In the cytoplasm, PER pools are small (low [PER]), and therefore its influence on CK1ε phosphorylation of substrates is minimal. On the other hand, once the [PER] is elevated by translocation to the nucleus, PER now exerts a considerable effect on CK1ε activity—inhibiting the phosphorylation of substrates such as P53 (output pathway) while enhancing the phosphorylation of key TTFL components such as CRY and PER itself (Figures 1 -4; Eide et al., 2002; Isojima et al., 2009). Indeed, CK1ε is known to associate rhythmically with CRYs, PERs, and P53 (Kategaya et al., 2012). Moreover, the concept that concentrations of clock proteins cycle by translocations within cells (and not just by transcription/translation) has been dramatically demonstrated recently for bacterial clocks (Cohen et al., 2014).
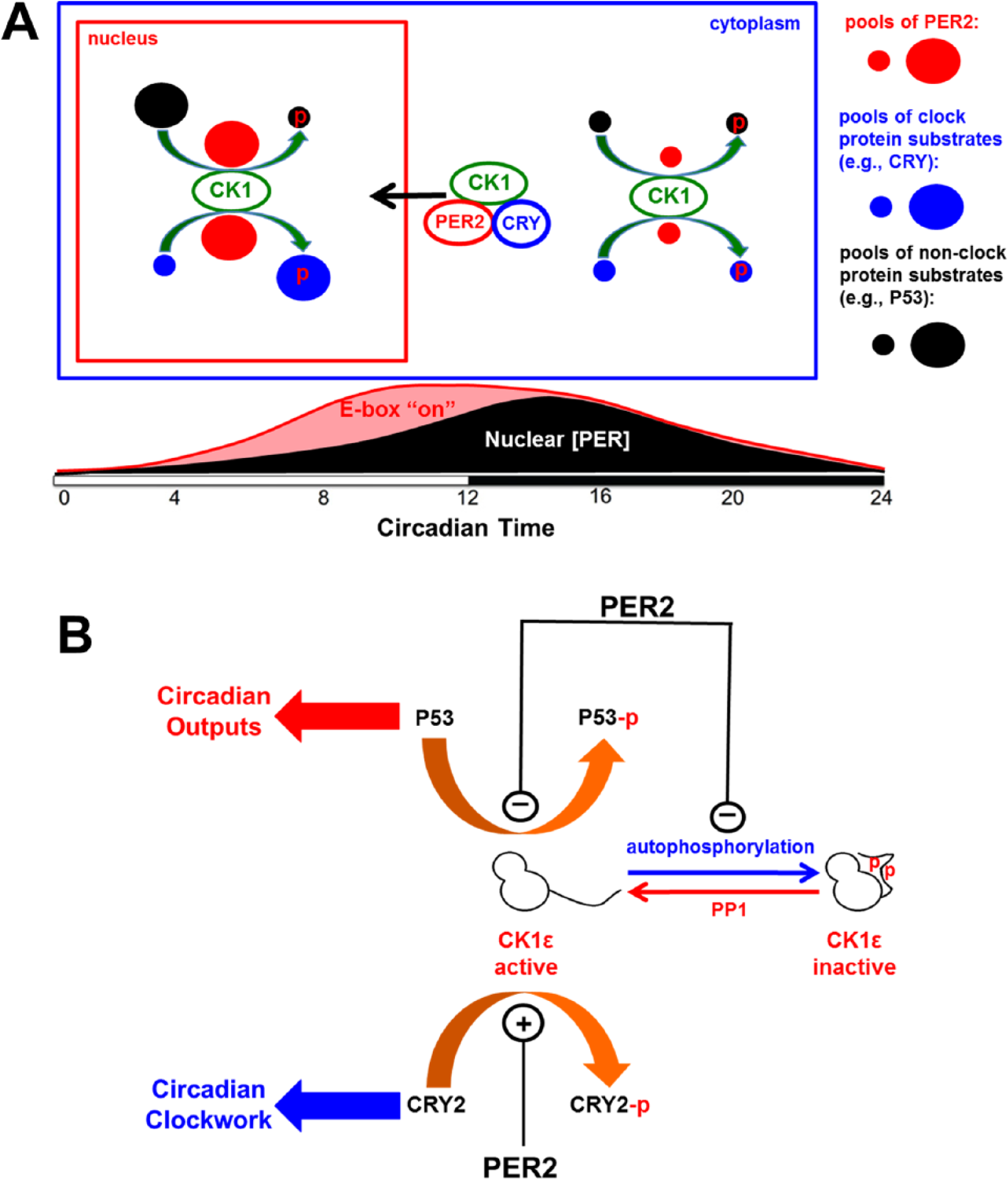
PER2 reciprocally regulates CK1ε-mediated phosphorylation events. (A) As the concentration pool of PER2 in the nucleus increases, it inhibits CK1ε-mediated phosphorylation on clock output substrates (e.g., P53) as indicated by the small pool of phosphorylated P53 in the nucleus (phosphorylation indicated by the red “p”). Simultaneously, PER2 stimulates CK1ε-mediated phosphorylation on substrates involved in the central clock mechanism (e.g., CRY). The lower portion of the panel shows the temporal changes of the PER2 pool in the nucleus and the correspondence of (PER2) with the repression of E-box transcription. (B) PER2 inhibits CK1ε-mediated phosphorylation of P53 (output) and CK1ε autoinhibition by autophosphorylation on its C-terminal domain (thereby maintaining CK1ε activity). At the same time, PER2 stimulates CK1ε-mediated phosphorylation of central clock proteins such as CRY.
Our results also illuminate the perplexing observation that CK1ε autoinhibits itself quickly by autophosphorylation in vitro (Graves and Roach, 1995; Cegielska et al., 1998; Rivers et al., 1998). The presence of PER2 suppresses this autoinhibition, thereby permitting continued CK1ε activity on its substrates (Figure 3). The presence of PER2 also enhances the activity of the kinase domain of CK1ε, which increases the phosphorylation level of the clock substrate CRY2 (Figure 4). The action on non–clock substrates (e.g., P53) is more complex—PER2 inhibits both the autoinhibition of CK1ε, but it also acts as an inhibitor of the phosphorylation of the substrate (Figure 6B). This inhibition effect is not dependent on the C-terminal domain of CK1ε, since the IC50 is similar for both CK1ε-FL and CK1ε-ΔC. The differential effects of PER2 on CK1ε activity on clock versus non–clock substrates may be due to reciprocal interaction between a PER2•CK1ε complex to clock versus non–clock proteins. In Neurospora, FRQ interacts with CK1 and was found to promote CK1-mediated phosphorylation of WC proteins by recruiting CK1 to the WC complex (He et al., 2006; the WC complex would be an example of clock-component proteins). The differential effects of PER2 on CK1ε-mediated phosphorylation of core clock proteins versus non–clock proteins may be due to physical attraction of PER2 for core clock proteins. These interactions could allow PER2 to function as a substrate-recruiting subunit of a PER2•CK1ε complex for the core clock proteins, so that CK1ε-mediated phosphorylation is promoted for clock components. On the other hand, a tight interaction between PER2 and CK1ε in a PER2•CK1ε complex might nonspecifically inhibit phosphorylation of non–clock proteins.
While continued activity of CK1ε is usually interpreted on the basis of phosphatases (e.g., PP1) that dephosphorylate CK1ε in vivo (Rivers et al., 1998; Cegielska et al., 1998), our data show another mechanism by which native CK1ε is active in vivo—there are likely to be a cadre of proteins that modulate CK1ε activity by inhibiting its autophosphorylation and enhancing its kinase activity toward clock substrates. PER2 is apparently a member of this cadre, and its significance in terms of circadian timing is that PER2’s effects will be circadian phase and compartment specific, where PER2’s maintenance of CK1ε activity will likely be relevant only in the nucleus and in the circadian phases when nuclear (PER) concentration is high (Figure 6A). In addition, because PER concentration in the nucleus is rhythmic, it will rhythmically control CK1ε phosphorylation on substrates (e.g., P53) that drive clock-regulated outputs (Figure 6B). A provocative interpretation of this result is that the role of the TTFL is to regulate rhythmic outputs by cyclically controlling CK1ε activity and not the usual interpretation that the role of CK1ε is to regulate the TTFL, which drives rhythmic gene expression. This concept is similar to the idea that the TTFL might drive daily activation of cAMP signaling that in turn regulates clock outputs (O’Neill et al., 2008). A less provocative and perhaps more accurate interpretation is that transcription/translation loops mutually cooperate with posttranslational loops to achieve a synergistic collaboration that orchestrates circadian timing and clock-controlled outputs.
Even though PERs and CRYs are usually considered corepressors of the circadian TTFL (Gekakis et al., 1998; Shearman et al., 2000; Reppert and Weaver, 2002), Ye and coworkers recently reported that CRY is the primary repressor in the TTFL and that PER alone has no effect on CLOCK/BMAL1-activated transcription (Ye et al., 2014). However, in the presence of CRY, they found that nuclear entry of PER acts upon CRY to displace CLOCK-BMAL1 from E-box promoters, thereby repressing transcription. It is tempting to conclude that reciprocal PER-modulated CK1ε-mediated phosphorylation of clock proteins such as PER, CRY, and BMAL1 (Eide et al., 2002) underlies the displacement/inactivation of CLOCK/BMAL1-activated transcription, especially since the CK1-binding domain of PER2 is necessary (but remarkably, not the PAS interaction domain!) for the essential repressive activity of E-box complexes (Ye et al., 2014). Moreover, we extend the initial findings of Isojima and coworkers (2009) to discover that CK1ε-mediated activity is temperature compensated on physiological clock substrates (full-length PER2) but temperature sensitive on key physiological output pathway proteins such as P53 (Figure 5). These data indicate that CK1ε-mediated phosphorylation is temperature compensated when its rate is involved in determining circadian period (Isojima et al., 2009; Lee et al., 2011), as on key clock protein substrates such as PER2, but it is not temperature compensated on non–clock protein substrates where the precise rate is not critical (e.g., the clock-regulated pathway exemplified by P53). How the phosphorylation reaction of CK1ε can be temperature compensated on one substrate while not compensated on another substrate is unknown, but presumably the 3-dimensional structures of the CK1ε/substrate interaction during the reaction differ between these 2 types of substrates, and these differences confer disparate thermodynamic properties.
In cyanobacteria, a circadian oscillation of protein phosphorylation can be reconstituted in vitro, indicating that an exclusively biochemical circadian oscillator is possible in the absence of a TTFL (Nakajima et al., 2005). Might such a biochemical oscillator exist for the mammalian circadian clock (Qin et al., 2010; Johnson, 2010; O’Neill and Reddy, 2011; Jolley et al., 2012)? If such a non-TTFL oscillator is present in mammalian cells, it must accommodate a constraint that is not present in cyanobacteria—namely, that the cycling compartmentalization of the key clock proteins alters stoichiometric relationships. For this reason, the biochemical timekeeper might not be a self-sustained oscillator as in cyanobacteria (Nakajima et al., 2005; Qin et al., 2010) but rather a temperature-compensated timer of a segment of the cycle in which clock proteins are at high concentration in the nucleus. Discussions of the mammalian circadian mechanism frequently identify the TTFL as providing essential feedback, but there is no adequate explanation for why this TTFL has a time constant as long as 24 h. Therefore, an amorphous “delay” is often introduced to justify how the mammalian clock can have such a long period, and the phosphorylation rate of clock proteins has been proposed as a possible candidate to provide this temperature-compensated delay. Based on the data reported here, we propose that a major component of the mammalian circadian mechanism that complements the TTFL is a reciprocal interaction between CK1δ/ε and PER that comprises a biochemical timer (Figure 6). This timer provides a temperature-compensated segment of the circadian cycle and partially establishes the long time constant of the clock.
Footnotes
Acknowledgements
We are grateful to the following for materials: Dr. David Virshup (pRSET-CK1ε), Dr. Steven Reppert (mPer2 cDNA), and Dr. Aziz Sancar (mCry2 cDNA). We appreciate the suggestions of Dr. Aziz Sancar on an earlier version of this manuscript. This research was supported by grants from the National Institutes of Health (R21HL102492, R01GM107434, and R01GM088595).
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.