
Abstract
The large repertoire of circadian rhythms in diverse organisms depends on oscillating central clock genes, input pathways for entrainment, and output pathways for controlling rhythmic behaviors. Stress-activated p38 MAP Kinases (p38K), although sparsely investigated in this context, show circadian rhythmicity in mammalian brains and are considered part of the circadian output machinery in Neurospora. We find that Drosophila p38Kb is expressed in clock neurons, and mutants in p38Kb either are arrhythmic or have a longer free-running periodicity, especially as they age. Paradoxically, similar phenotypes are observed through either transgenic inhibition or activation of p38Kb in clock neurons, suggesting a requirement for optimal p38Kb function for normal free-running circadian rhythms. We also find that p38Kb genetically interacts with multiple downstream targets to regulate circadian locomotor rhythms. More specifically, p38Kb interacts with the period gene to regulate period length and the strength of rhythmicity. In addition, we show that p38Kb suppresses the arrhythmic behavior associated with inhibition of a second p38Kb target, the transcription factor Mef2. Finally, we find that manipulating p38K signaling in free-running conditions alters the expression of another downstream target, MNK/Lk6, which has been shown to cycle with the clock and to play a role in regulating circadian rhythms. These data suggest that p38Kb may affect circadian locomotor rhythms through the regulation of multiple downstream pathways.
Circadian clocks are conserved molecular processes that dictate the daily behavioral rhythms of organisms and play an integral role in the ability of organisms to respond to environmental stimuli and regulate metabolism (Reppert and Weaver, 2002). In Drosophila, the core clock is mediated by the period (per) and timeless (tim) genes, which are under the control of the CLOCK/CYCLE (CLK/CYC) transcription factor heterodimer. During the day, per and tim transcript levels increase, with protein levels peaking in early night. The PER and TIM proteins then accumulate and dimerize in the cytoplasm, with both PER and TIM becoming phosphorylated. Around midnight, the PER and TIM proteins then translocate into the nucleus and bind to CLK/CYC, thus repressing their own transcriptional activation by late night. PER continues to be phosphorylated in the nucleus, leading to a hyperphosphorylated state that then triggers degradation of the PER protein during early day, relieving the repression of CLK/CYC and now allowing for transcription of the per and tim loci (reviewed in Allada et al., 1998). The cycling of these proteins dictates the circadian rhythms of the organism, with mutations in these genes leading to shortened or lengthened circadian rhythms or even arrhythmia (reviewed in Hardin and Panda, 2013).
The circadian clock dictates metabolic rates over the course of a day-night cycle (Langmesser and Albrecht, 2006). Interestingly, metabolism is closely linked with oxidative stress caused by reactive oxygen species (ROS). In Drosophila, the levels of oxidative stress cycle throughout the 24-h day, reaching peak levels during the subjective day and declining during the subjective night (Krishnan et al., 2008). Furthermore, Krishnan et al. (2008) found that sensitivity to the oxidizing agent hydrogen peroxide is dependent on the time of day that the animals are exposed, with increased mortality at night (ZT20, 8 h after lights off). Furthermore, oxidative stress levels fail to cycle in per mutants, which are also more susceptible to exposure to oxidizing agents (Krishnan et al., 2008). Treatment with another oxidizing agent, paraquat, particularly affects the clock in peripheral tissues, resulting in a dampening of per transcription cycles caused by repression of CLK activity (Zheng et al., 2007). In mice, loss of the CYC homolog BMAL1 also leads to increased ROS (Kondratov et al., 2006). In addition, the levels of the antioxidant enzyme glutathione (GSH) cycle in the fly, and this cycling is lost in per and cyc mutants (Beaver et al., 2012).
Disruptions in the circadian clock by either mutation or environmental factors can lead to a reduced lifespan in many organisms and pathological outcomes in humans such as cancer, cardiovascular disease, metabolic syndromes, and age-associated brain dysfunction (Kondratova and Kondratov, 2012), suggesting a role for circadian rhythms in aging. As organisms age, circadian and sleep rhythms deteriorate (Froy, 2011; Joshi et al., 1999; Ko et al., 2002; Rezaval et al., 2008; Zhdanova et al., 2011; Zheng et al., 2007). This age-dependent deterioration is accompanied by decreased expression of clock genes, such as per (Krishnan et al., 2009). Interestingly, in free-running conditions (constant darkness), as flies age (by midlife ~35 days old) arrhythmia increases as does the length of free-running periods (Rakshit et al., 2012; Umezaki et al., 2012). Furthermore, loss of clock regulatory genes such as per or cyc leads to premature aging in mice and flies as well as an increase in age-dependent disease-like phenotypes (Hendricks et al., 2003; Koh et al., 2006; Kondratov et al., 2006; Krishnan et al., 2009; Turek et al., 1995a; Turek et al., 1995b; Umezaki et al., 2012). The neurotransmitter pigment-dispersing factor (PDF) decreases with age, and overexpression of PDF in clock neurons rescues age-dependent circadian rhythm defects (Umezaki et al., 2012). In addition, the longevity gene Foxo has been shown to play a role in circadian rhythms, as Drosophila foxo mutants have accelerated age-dependent deterioration in circadian rhythms (Zheng et al., 2007).
We have recently found that the stress response protein kinase p38 MAP Kinase (p38K) is a regulator of lifespan and aging in the fly (Vrailas-Mortimer et al., 2011). The activity of p38K is regulated by dual phosphorylation of the TGY motif, and in a variety of organisms the phosphorylation state of p38K cycles throughout the 24-h day (Chik et al., 2004; Hayashi et al., 2003; Pizzio et al., 2003; Vitalini et al., 2007). In Neurospora, p38K phosphorylation cycles through the day with peak levels in the early morning (Vitalini et al., 2007). Similarly, in the rat pineal gland, phospho-p38K levels increase throughout the night and are sensitive to light exposure (Chik et al., 2004). However, in hamster suprachiasmatic nuclei, p38K phosphorylation increases during the day, and light exposure during the dark induces phosphorylation (Pizzio et al., 2003). In contrast, phospho-p38K in the chick pineal gland doesn’t cycle; however, inhibition of p38K during the day in the pineal gland leads to a lengthening of the free-running period of the melatonin secretion rhythm (Hayashi et al., 2003). Interestingly, inhibition of per1 in rat spinal cord astrocytes leads to increased p38K phosphorylation (Sugimoto et al., 2014), while loss of per in flies leads to a decrease in p38K mRNA expression (Kula-Eversole et al., 2010). Circadian studies on p38K have been limited in vertebrate systems due to the presence of multiple p38K genes and the lack of a viable p38Kα (the primary mouse p38K gene) knockout mouse (Adams et al., 2000). Therefore, we tested the role of p38K in circadian rhythms using the fruit fly Drosophila melanogaster, which has two p38K genes, p38Ka and p38Kb. Our observations suggest that p38Kb is a regulator of circadian rhythms, since inhibition of p38Kb results in a significantly longer cycling period under free-running conditions but normal rhythms in the presence of light cues. We find that p38kb genetically interacts with per to regulate circadian rhythms. Furthermore, we find that p38Kb may also regulate circadian rhythms through the additional downstream targets Mef2 and MNK/Lk6. Thus, p38K signaling may act as a node to relay upstream inputs to a variety of downstream effectors to precisely regulate circadian rhythms.
Materials and Methods
Fly Stocks, Rearing, and Transgenics
Flies were raised at 25 °C in a 12-h:12-h light-dark (LD) cycle on standard cornmeal molasses media. p38KbΔ45, p38KbΔ25, Δp38Ka, UAS-p38Kb wt, UAS-p38Kb ala2, UAS-p38Kb KD3-FLAG, UAS-p38Kb KD8-FLAG, and p38Kb-GAL4 were as described previously (Vrailas-Mortimer et al., 2011). We obtained per0, Pdf-GAL4, Cry-GAL4, and UAS-nLacZ from the Bloomington Stock Center. Pdf 0.5-GAL4 and UAS-Mef2-En were a gift from Justin Blau. The UAS-p38Kb Asp transgene was created through site-directed mutagenesis that replaced Thr183 and Tyr185 with Asp. This mutated transgene was then cloned into pUAST and used to generate transgenic Drosophila (BestGene Inc.). The p38K double-knockout (DKO) flies (p38KbΔ25; Δp38Ka) were isogenized by outcrossing to a w1118 background as the original stock was found to have the perSLIH mutation.
Measurement of Circadian Rhythms
For analysis of p38K mutants, either male or virgin female flies 1 to 2 days old were placed individually in Drosophila activity monitors (DAM, Trikinetics, Waltham, MA) and entrained in a 12-h:12-h LD cycle for 3 days and then switched to 3 days of constant darkness. To verify that changes in circadian rhythm behavior are not due to impaired locomotor activity, virgin female flies 1 to 2 days old were entrained in a 12-h:12-h LD cycle, then placed in the monitors for 3 days of constant darkness, and then switched to a 12-h:12-h LD cycle for 3 days. For transgenic analysis, virgin female flies 1 to 3 days old were entrained in a 12-h:12-h LD cycle and then placed in the monitors for 6 days of constant darkness. For aging experiments, flies were aged for 13 to 15 days at 25 °C in a 12-h:12-h LD cycle and then placed in the monitors for 6 days of constant darkness. Raw data of locomotor activity (light beam crosses) were collected in 1-min bins and plotted as actograms using the ImageJ plugin, ActogramJ (Schmid et al., 2011). Alternatively, raw data were collected in 30-min bins and plotted as the average number of beam breaks over the 6 days of constant darkness. Data acquired from the DAM were analyzed for the length of the period (tau, τ) and regularity and strength of rhythms (rhythmicity index, RI). Maximum entropy spectral analysis (MESA) (Dowse, 2009; Levine et al., 2002) was used to estimate τ. The presence of significant rhythmicity was verified by autocorrelation analysis (Chatfield, 1989; Dowse, 2009; Levine et al., 2002). The presence of 2 or more autocorrelation peaks exceeding the 95% confidence interval (2/√N) corresponding to a spectral peak was taken as evidence of a robust rhythm in the data set. The regularity of the rhythm was assessed by further interpretation of the autocorrelation function. The height of the third peak, counting the one at lag zero as #1, is the RI (Dowse, 2009; Levine et al., 2002). Since fly rhythms are often bimodal, we counted only the peaks occurring in approximately 24-h windows to establish RI. Drosophila locomotor activity rhythms are often complicated by high frequency noise. To make interpretation of the actograms simpler and to produce cleaner spectra, the data were filtered using a 2-pole low-pass Butterworth filter with a 4-h cutoff (Dowse, 2009; Hamming, 1983; Levine et al., 2002). For the actograms, the data were run through the filter twice, once in a forward direction and the other time in reverse, as the iterative nature of the filter induces a 4-h phase delay, which is negated by the second passage. As multiple filtering can create artifact visible in subsequent analysis (Hamming, 1983), the data operated on by the MESA program were filtered only once, as any phase shift is immaterial.
Western Immunoblot Assay
Flies were reared in a 12-h:12-h LD cycle and collected during the second day of entrainment at ZT1, ZT7, ZT13, and ZT19 or placed in constant darkness and collected during the second day of constant darkness at CT1, CT7, CT13, and CT19. Thirty heads from male flies were homogenized in EB1 as described by Chiu et al. (2011) and in 2× Laemmli buffer. Equivalent amounts of each sample were loaded on a 4% gel. Proteins were transferred to PDVF membranes using the manufacturer’s protocols. Antibodies used were rabbit anti-PER (a gift from A. Sehgal, 1:40,000) and rabbit anti-SERCA (Sanyal et al., 2005 1:10,000). Blots were developed using enhanced chemiluminescence (ECL Plus; Pierce/Thermo Scientific) and scanned using a flatbed scanner. All immunoblots were performed in triplicate.
Co-Immunoprecipitation
Flies were reared in a 12-h:12-h LD cycle and collected at ZT1. We expressed 2 copies of p38Kb KD (UAS-p38Kb KD3-FLAG and UAS-p38Kb KD8-FLAG) in clock neurons using Cry-GAL4. Cry-GAL4 outcrossed to w1118 was used as a genetic background control. Thirty heads from males per genotype were collected and homogenized in high salt buffer as described previously (Mortimer and Moberg, 2007). p38Kb KD was immunoprecipitated on anti-FLAG M2 beads (Sigma-Aldrich) and eluted using 3× FLAG peptide (Sigma-Aldrich). Immunoprecipitated samples were run on a 4% gel. Proteins were transferred to PDVF membranes using the manufacturer’s protocols. Antibodies used were mouse anti-FLAG M2 (Sigma, 1:5000) and rabbit anti-PER (a gift from A. Sehgal, 1:20,000). Blots were developed using ECL Plus.
Immunohistochemistry
For analysis of p38Kb expression, flies were reared in a 12-h:12-h LD cycle and collected at ZT2 for PDF costaining and ZT16 for PER costaining. In Lk6 expression analysis, flies were reared in a 12-h:12-h LD cycle and then reared in constant darkness and collected on the second day at CT1, CT7, CT13, and CT19. Flies collected at various time points were fixed in 4% paraformaldehyde at 4 °C for 24 h. Fixed brains were dissected in phosphate-buffered saline; permeabilized in phosphate-buffered saline plus 0.1% Triton X-100 (PBT) for 30 min; blocked in 0.15% Triton X-100, 2% BSA, and 5% NGS (block) for 1 h; and stained overnight in primary antibody solution in block. All washes were carried out in PBT. After secondary antibody staining in block, brains were mounted in Vectashield (Vector Laboratories) and imaged with a Zeiss 510 laser scanning confocal microscope. Lk6 stainings were imaged on the same settings for all genotypes at all time points. Antibodies used were rabbit anti-PER (gift from Amita Sehgal), mouse anti-PDF (DSHB), rabbit anti-Lk6 (gift from Jordan Raff), rabbit anti-βgal (Gibco), and mouse anti-βgal (Gibco).
Results
p38Kb Is Expressed in Clock Neurons
There are two p38K homologs in Drosophila, p38Ka and p38Kb, which are nearly identical in their amino acid sequence, and a number of previous studies have shown partial redundancy between these 2 genes (Chen et al., 2010; Vrailas-Mortimer et al., 2011). For instance, null mutations in either p38Ka or p38Kb are viable as adults (although p38Kb mutants have a shortened life span; Vrailas-Mortimer et al., 2011), while complete knockout of both genes is lethal. Due to the lack of p38Ka and p38Kb specific antibodies, we generated a p38Kb transcriptional reporter line (Vrailas-Mortimer et al., 2011), in which the endogenous p38Kb promoter drives the expression of a UAS-nLacZ reporter transgene. As we previously described, p38Kb is expressed in neurons throughout the adult brain (Fig. 1A and Vrailas-Mortimer et al., 2011) and is specifically expressed in a subset of clock neurons: the large ventral lateral neurons (I-LNv) as marked by PER and PDF expression (Fig.1, A-I) and in the PER positive DN3 neurons (Fig.1, J-L). These data are consistent with microarray studies which found that both p38Ka and p38Kb are expressed in PDF positive neurons (Kadener et al., 2009; Kula-Eversole et al., 2010; Nagoshi et al., 2010; Wijnen et al., 2006). Interestingly, these studies also found that neither p38Ka nor p38Kb transcripts cycle (Kadener et al., 2009; Kula-Eversole et al., 2010; Nagoshi et al., 2010; Wijnen et al., 2006), suggesting that if the p38K genes are under circadian regulation, it may be through phosphorylation of the proteins. In a previous study, we used mutational analysis to detect differences in p38Ka and p38Kb protein expression. While we found that very low levels of total p38Ka and undetectable levels of phospho-p38Ka were expressed in whole heads, we found that phospho-p38Kb is highly abundant, suggesting that p38Kb may be the primary p38K gene for neuronal function (Vrailas-Mortimer et al., 2011). Dusik et al. (2014) reported that both p38Ka and p38Kb were found to be expressed in clock neurons, and phosphorylation of both proteins was found to cycle in a subset of these neurons. These data suggest that both p38Ka and p38Kb may play a role in clock neurons.
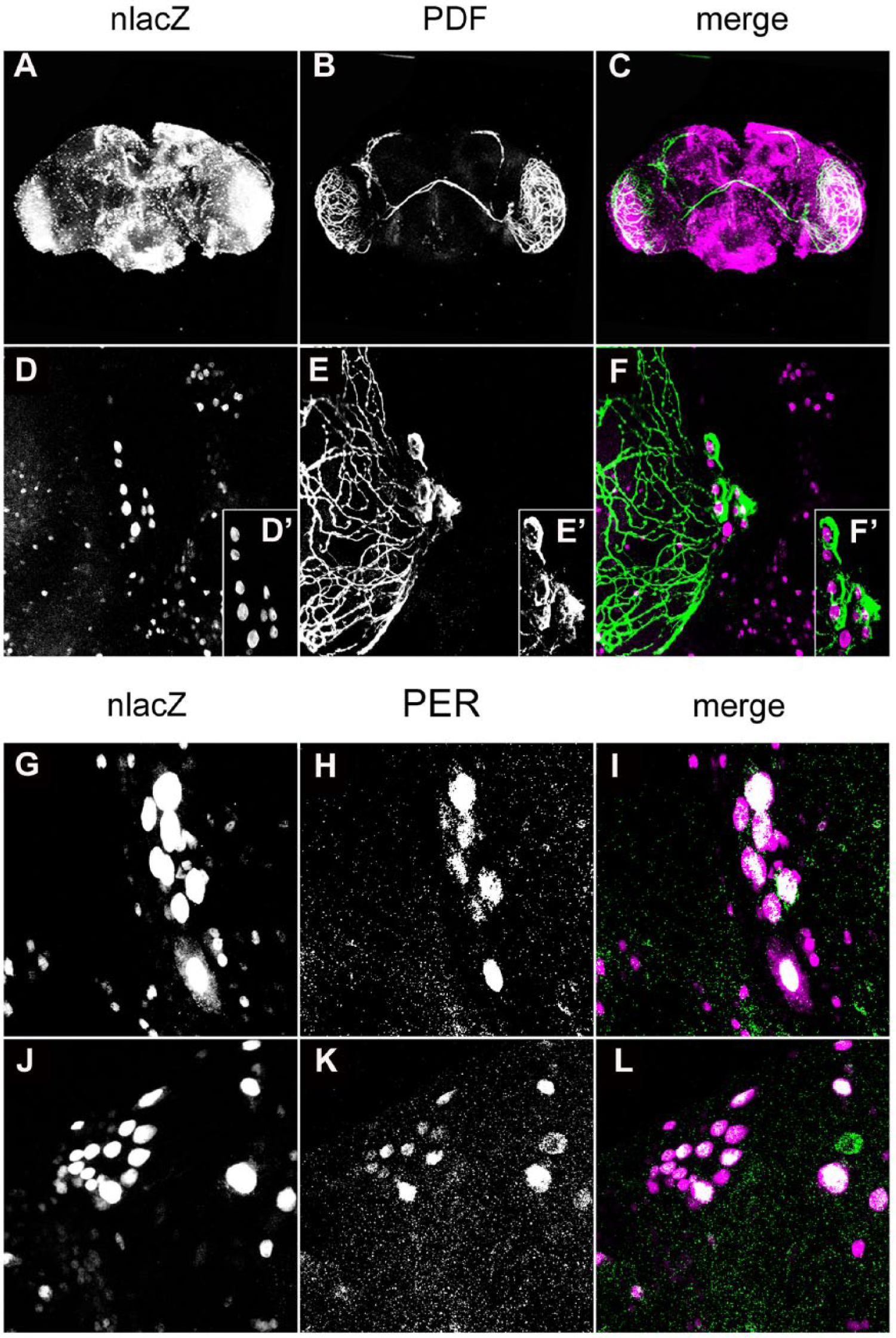
p38K is expressed in clock neurons in adult Drosophila brains. A reporter of p38Kb transcriptional expression, p38Kb-GAL4:UAS-nLacZ (A, D, G and J, magenta in C, F, I and K) is expressed in PDF (B and E, green in C and F) and PER (H and green in I) positive I-LNv neurons and PER positive DN3 neurons (K and green in L).
p38K Mutants Have Aberrant Circadian Rhythms
Since p38Ka and p38Kb are expressed in clock neurons (Fig. 1 and Kadener et al., 2009; Kula-Eversole et al., 2010; Nagoshi et al., 2010; Wijnen et al., 2006), we tested whether p38K signaling plays a role in regulating circadian rhythms using a locomotor-based assay in which the animals display a rhythmic locomotor behavior when entrained to a 12-h:12-h LD cycle. We first measured locomotor behavior rhythms in LD in single null mutants of either p38Ka (Δp38Ka) or p38Kb (p38KbΔ45) as well as a viable DKO combination (p38K DKO) of a null allele of p38Ka (Δp38Ka) and a hypomorphic allele of p38Kb (p38KbΔ25) and found no effect on locomotor behavior (data not shown). Therefore, we tested whether loss of p38K under free-running conditions (constant darkness [DD]) affects circadian locomotor behavior. We find that loss of either p38Ka or p38Kb does not affect normal circadian rhythms in young animals. These findings are consistent with the recent results from Dusik et al. (2014). Interestingly, we find that a significantly larger proportion of p38K DKO animals are arrhythmic (Fig. 2, A and C; Table 1; and Suppl. Fig. S1), while animals that are not arrhythmic display normal circadian rhythms. This dichotomy between normal and abnormal rhythms is not entirely unexpected as the p38K DKO phenotypes are partially penetrant (Vrailas-Mortimer et al., 2011). In addition, we find that 32% of p38K DKO animals also display ultradian rhythms compared with 4% of wild-type controls (Fig. 3).
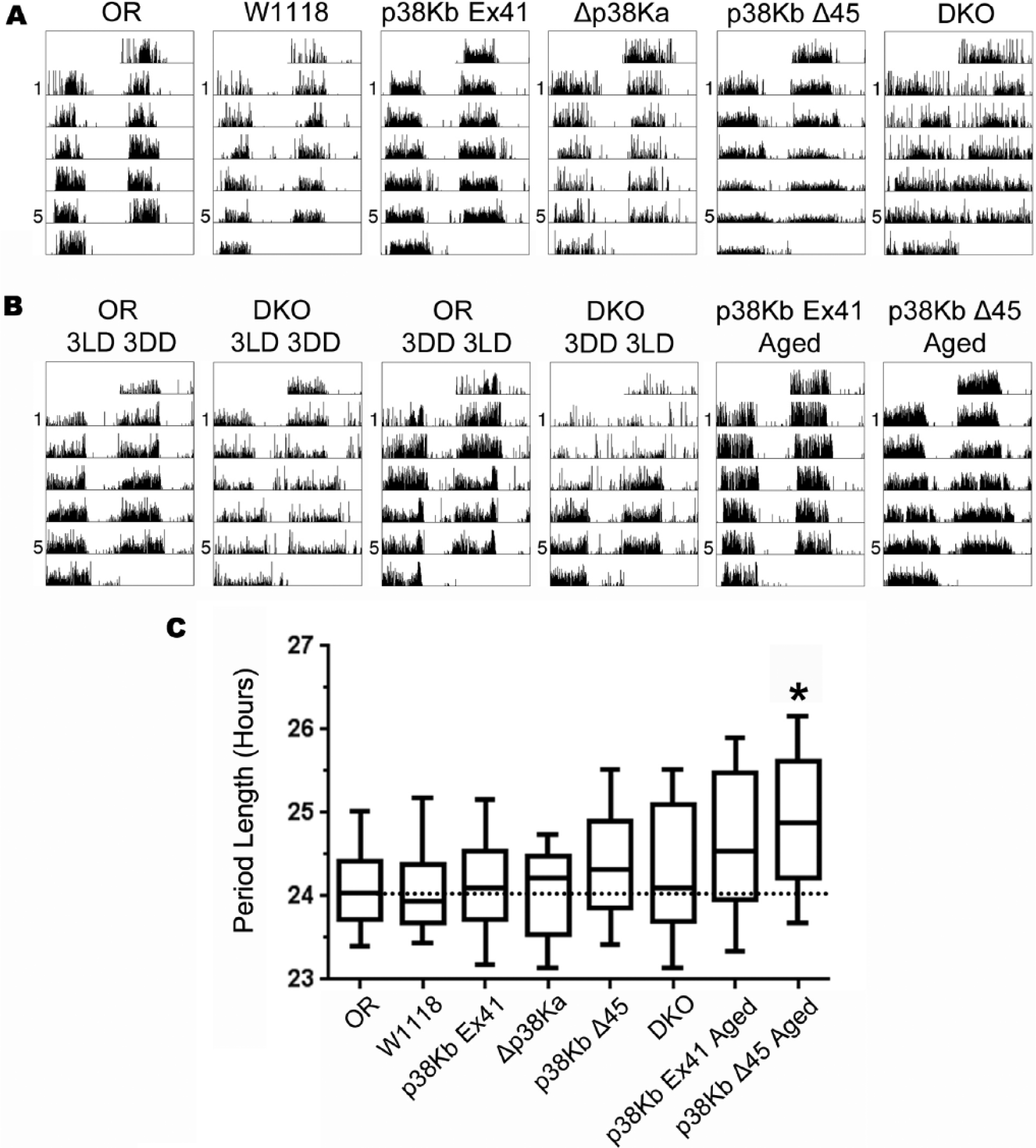
p38K mutants have longer free-running rhythms. (A) p38K single mutants exhibit normal circadian rhythms. However, loss of both p38K genes (DKO) results in mostly arrhythmic animals. (B) p38K DKO circadian defect is not a result of age-dependent locomotor dysfunction. In addition, aged p38Kb single mutants (p38Kb Δ45) display an increase in period compared with age-matched controls (p38Kb Ex41). (C) Box and whisker plots of young and aged animals. No significant difference in tau for young mutants and controls was determined by ANOVA. Aged p38Kb mutants have a significant increase in period length compared with age-matched controls (p38Kb Ex41) using Student’s t test. *p < 0.05.
Loss of p38K results in increased arrhythmicity and increased tau with age.
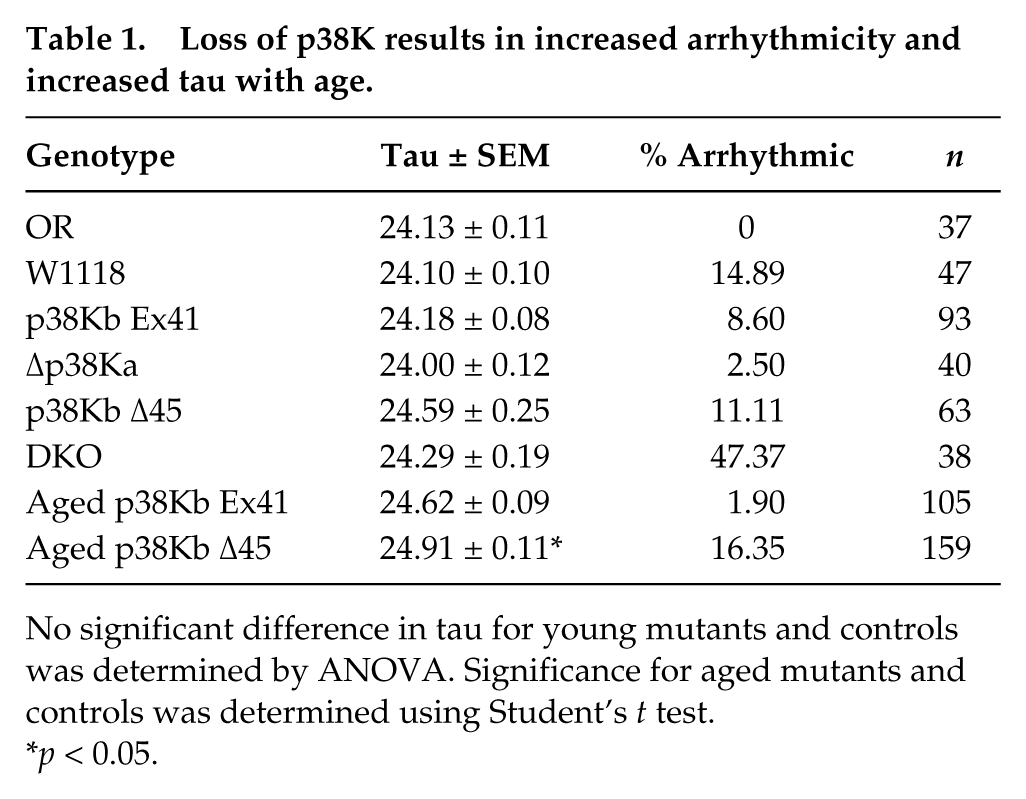
No significant difference in tau for young mutants and controls was determined by ANOVA. Significance for aged mutants and controls was determined using Student’s t test.
p < 0.05.
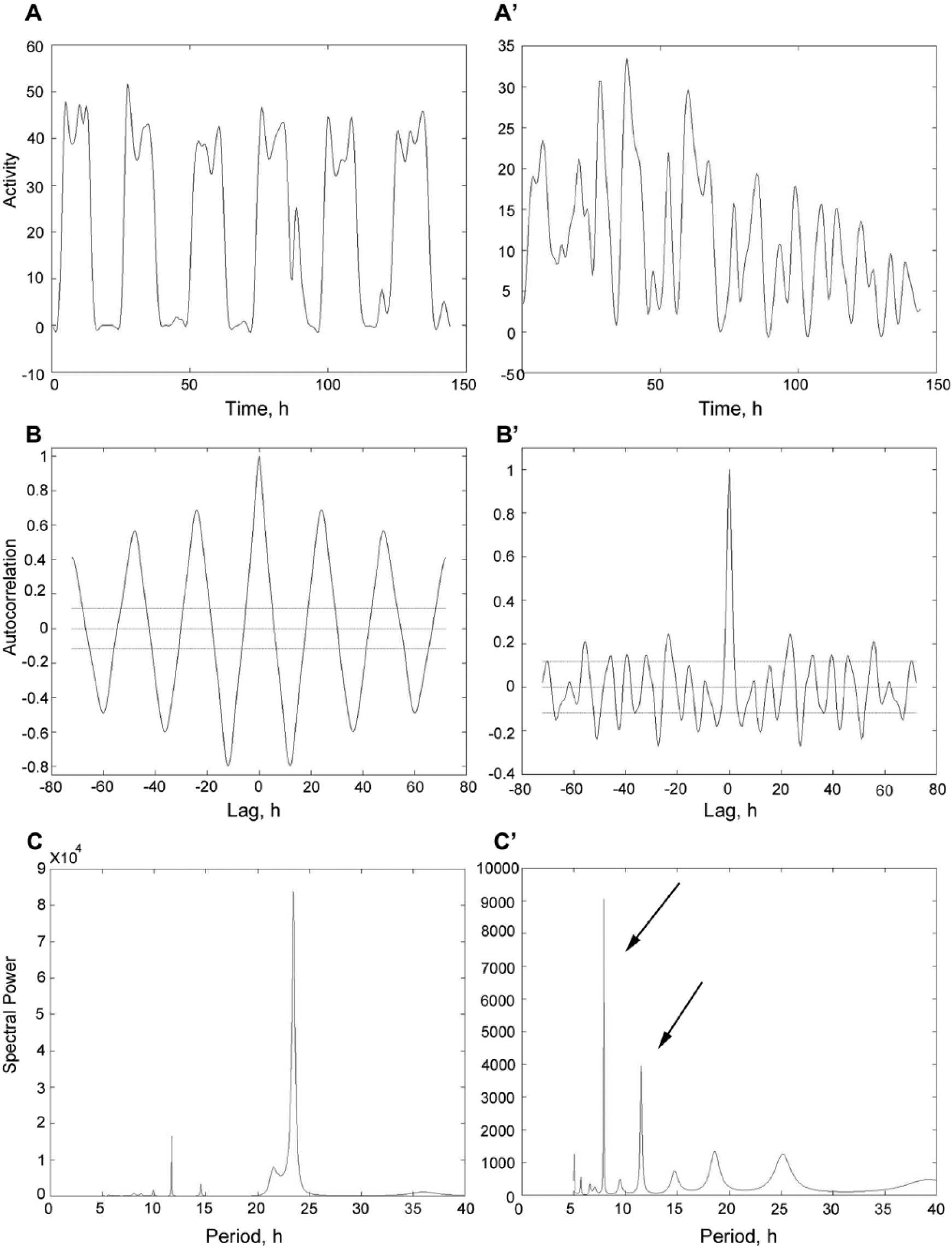
p38K mutants display ultradian rhythms. p38K DKO and precise excision animals were reared in 12-h:12-h light-dark cycle (LD) and then placed in constant darkness for 6 days. (A and A′) actograms, (B and B′) autocorrelograms, and (C and C′) MESA spectrum. (A-C) p38Kb Ex41 animals show predominantly normal circadian rhythms as depicted here. (A′-C′) In contrast, spectra of p38K DKO animals commonly reveal the appearance of ultradian rhythms (black arrows) as in this example. Compare C′ with C.
Since we have previously found that the p38K DKO animals have an age-dependent locomotor defect (Vrailas-Mortimer et al., 2011), we wanted to confirm that the increase in arrhythmicity in the p38K DKO animals is not the result of a locomotor deficit. As a control, animals were reared in a 12-h:12-h LD cycle and then transferred to constant darkness for 3 days (3LD, 3DD). Consistent with our earlier observation, these animals continue to display arrhythmic behavior only in DD. Since the 3DD comes at an age when p38K DKO animals display locomotor behavior defects, we placed LD-entrained p38K DKO animals in constant darkness for 3 days followed by a return to a 12-h:12-h LD cycle (3DD 3LD). Again, the p38K DKO animals were arrhythmic in DD, but locomotor behavior was restored when they were returned to LD (Fig. 2B), suggesting that p38K signaling is required for normal free-running circadian rhythms and any behavioral defects are not a consequence of impaired locomotor function. As we have found that p38Kb single mutants have several age-dependent phenotypes (Vrailas-Mortimer et al., 2011), we tested whether loss of p38Kb leads to an age-dependent loss of circadian rhythms. We found that aged p38Kb mutants have a modest but significant increase in tau of 24.9 h compared with a precise excision control (p38Kb Ex41) of 24.6 h (Fig. 2, B and C; Table 1; and Suppl. Fig. S1).
Optimal p38Kb Signaling Is Required in Clock Neurons for Maintaining Normal Circadian Rhythms
Because global knockout of p38Ka and p38Kb can lead to pleiotropic effects, we decided to test the role of p38Kb specifically in clock neurons. Therefore, we overexpressed 2 different dominant negative forms of p38Kb, both of which we have previously found to inhibit p38Kb signaling in vivo (Vrailas-Mortimer et al., 2011). The first is a phospho-null form of p38Kb (p38Kb ala2) in which the critical Thr and Tyr residues of the TGY phosphorylation motif have been mutated to Ala. Although p38Kb ala2 can interact with upstream kinases, it cannot be phosphorylated, thus preventing activation of p38Kb. Reminiscent of p38KbΔ45 mutants, overexpression of p38Kb ala2 in clock neurons using either a Pdf 0.5-GAL4 or a Cry-GAL4 driver results in a significant increase in arrhythmicity and tau in free-running conditions of 25.9 h and 25.2 h, respectively (Figs. 4 and 5, Tables 2 and 3, and Suppl. Figs. S2 and S3). Furthermore, overexpression of a second dominant negative construct, a kinase dead version of p38Kb (p38Kb KD) that cannot phosphorylate downstream targets, also resulted in an increased arrhythmicity and period under free-running conditions when used in a single copy (p38Kb KD3 [weak line] or p38Kb KD8 [strong line, data not shown]) or when used in 2 copies (p38Kb KD3 and p38Kb KD8, 25.3 h for expression with Pdf 0.5-GAL4 and 25.5 h for expression with Cry-GAL4) (Figs. 4 and 5, Tables 2 and 3, and Suppl. Figs. S2 and S3). These results further confirm that p38K signaling influences circadian rhythms and mirror recent findings that RNAi knockdown of p38Kb or expression of a different p38Kb dominant negative transgene (in which only 1 of the phosphorylation sites is mutated) in clock neurons also results in period lengthening under free-running conditions (Dusik et al., 2014).
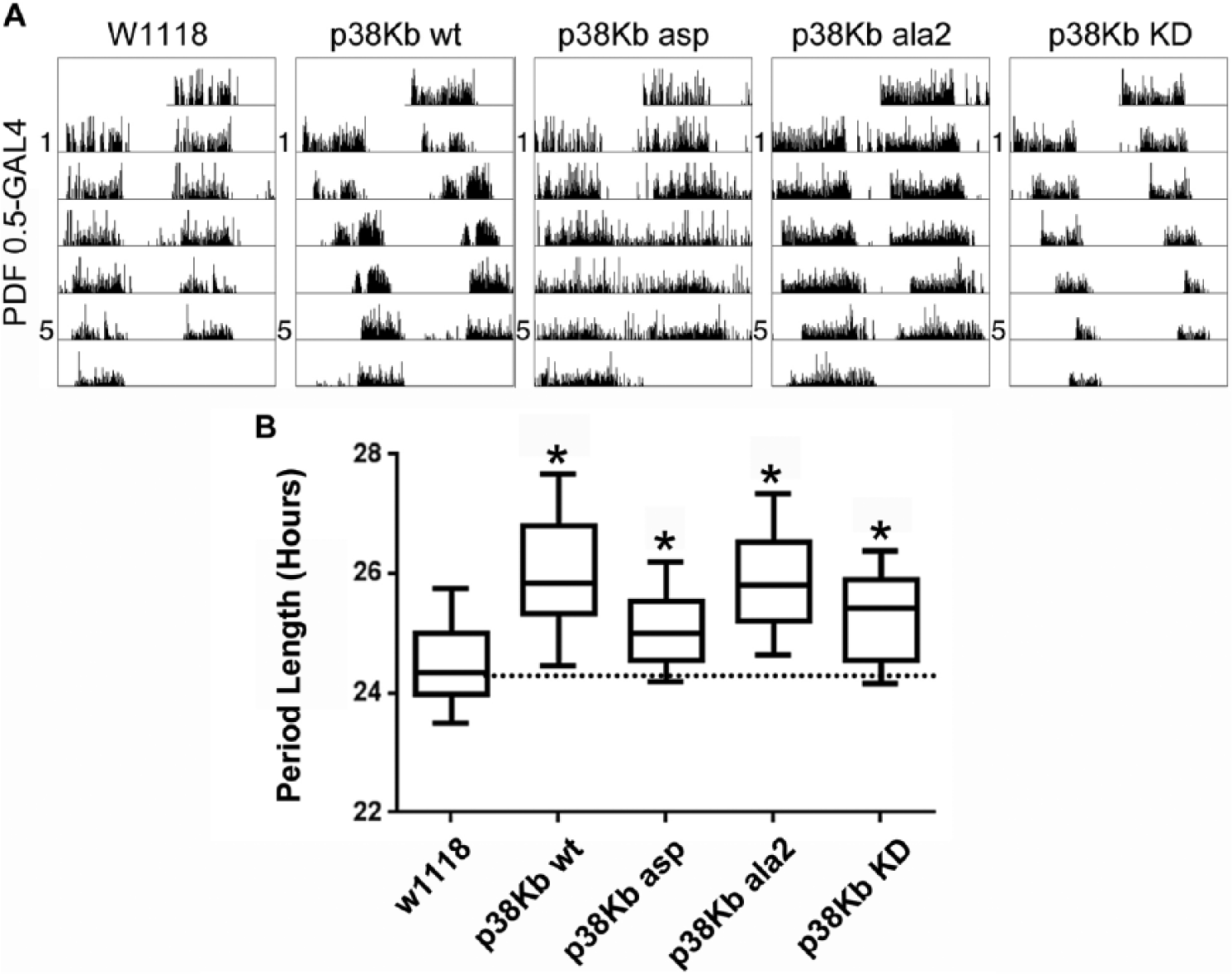
Either p38Kb activation or inhibition in clock neurons results in arrhythmic flies and lengthens the free-running rhythm. (A) Both activation (p38Kb wt and p38Kb asp) and inhibition (p38Kb ala2 and p38Kb KD) of p38Kb signaling in PDF-expressing clock neurons result in an increased period. (B) Box and whisker plots. Statistical significance was determined using a Dunnett’s test with all genotypes compared with the W1118 Pdf 0.5-GAL4 control. *p < 0.05.
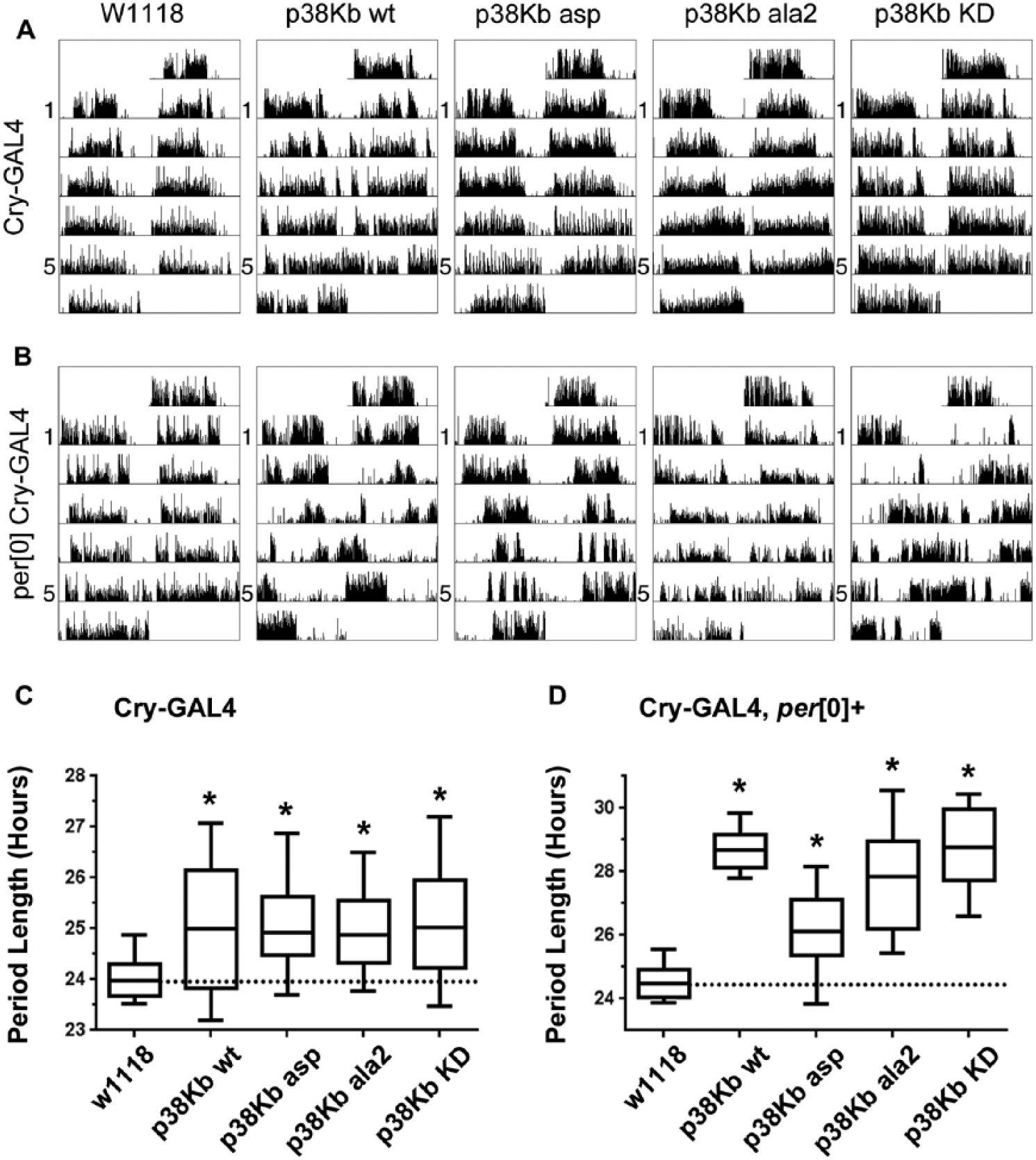
Reduced PER strongly enhances p38K-dependent circadian rhythm phenotypes. (A) Both activation (p38Kb wt and p38Kb asp) and inhibition (p38Kb ala2 and p38Kb KD) of p38K signaling in cry-expressing clock neurons result in an increased period. (B) Loss of a single copy of per results in decreased arrhythmicity but increased period for p38Kb wt overexpression animals and increased arrhythmicity in p38Kb inhibition backgrounds. (C) Box and whisker plots of the data in part A. Statistical significance was determined using Dunnett’s test with all genotypes compared with the W1118 Cry-GAL4 control. *p < 0.05. (D) Box and whisker plots of the data in part B. Statistical significance was determined using Dunnett’s test with all genotypes compared with the per[0]/+ Cry-GAL4 control. *p < 0.001.
Tau and arrhythmicity of p38K expression in Pdf-expressing clock neurons.
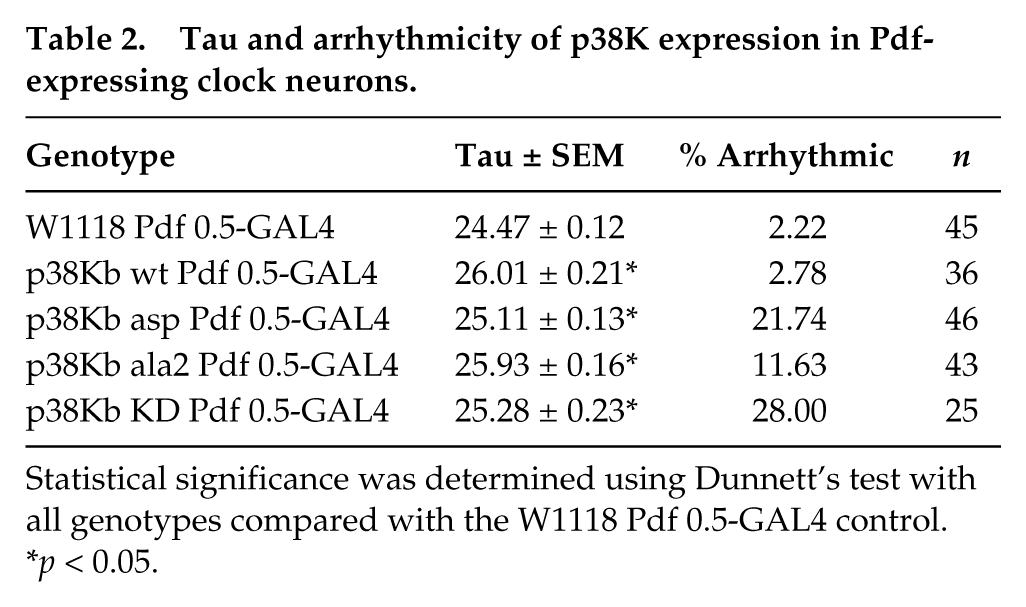
Statistical significance was determined using Dunnett’s test with all genotypes compared with the W1118 Pdf 0.5-GAL4 control.
p < 0.05.
Aberrant p38K signaling in cry-expressing neurons affects tau and arrhythmicity.
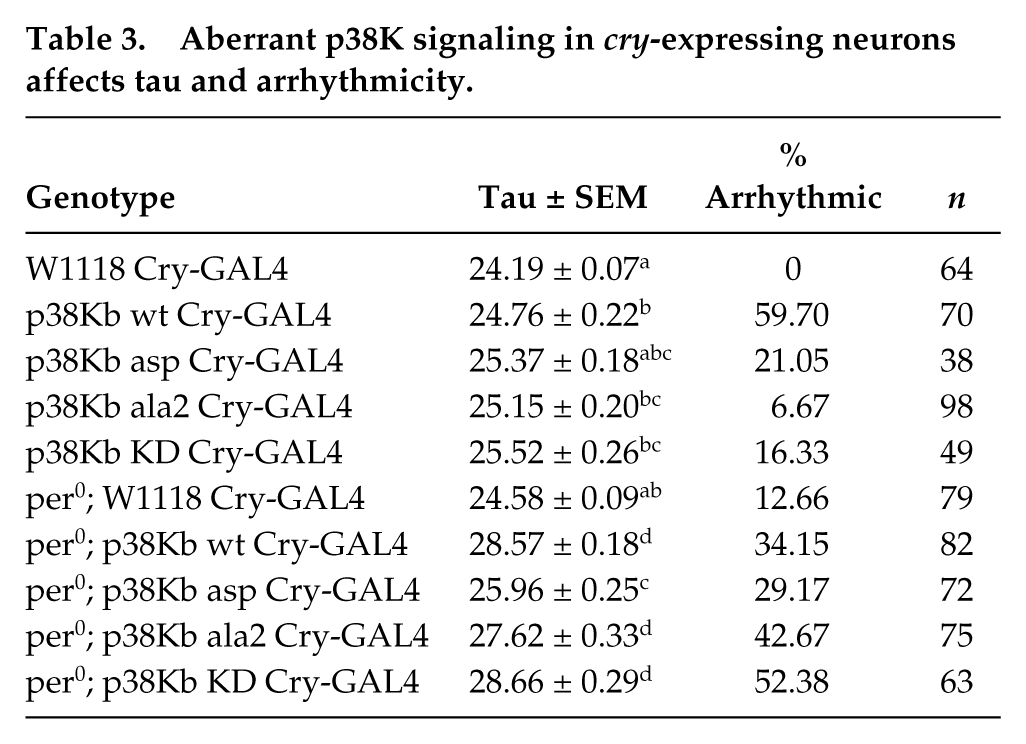
Significance between groups was determined by ANOVA followed by Tukey’s HSD. Significance groups are indicated by superscript letters.
Paradoxically, overexpression of wild-type (p38Kb wt) in clock neurons in free-running conditions also results in an increased tau as well as an increase in the number of arrhythmic animals (Figs. 4 and 5, Tables 2 and 3, and Suppl. Figs. S2 and S3), which also was observed by Dusik et al. (2014). As the phosphorylation of p38Kb is important for its downstream functions, we tested whether increasing the amount of activated p38Kb would result in a further increase in period length or arrhythmicity. We found that overexpression of a p38Kb phospho-mimic (p38Kb asp, in which the Thr and Tyr residues of the TGY motif are mutated to Asp) in PDF positive neurons under free-running conditions leads to a further increase in the number of arrhythmic animals compared with p38Kb wt overexpression (Fig. 4, Table 2, and Suppl. Fig. S2). However, p38Kb asp expression using the Cry-GAL4, which is more widely expressed, results in a further increase in period length compared with p38Kb wt overexpression (Fig. 5, Table 3, and Suppl. Fig. S2).
Our observations are contrary to the expectation that p38Kb signaling would act in a bidirectional manner, with overexpression and inhibition of p38Kb resulting in opposite phenotypes. In fact, our results point to a situation where normal clock function requires optimal p38Kb signaling. Accordingly, different magnitudes or spatial patterns of p38Kb activation may result in different outcomes on circadian rhythms, an idea consistent with the recent observation that the levels of p38K phosphorylation cycle in specific clock neurons (Dusik et al., 2014).
p38Kb-Mediated Regulation of Circadian Rhythm Is Sensitive to Levels of Per
Since aberrant p38Kb signaling leads to an increase in tau only in free-running conditions, much like loss of the core clock component per, we hypothesized that p38Kb may genetically interact with per. We find that loss of a single copy of per (per0, which when homozygous results in arrhythmia in free-running conditions) in the p38Kb wt overexpression background rescues the p38Kb wt arrhythmicity phenotype such that a smaller percentage of flies of this genotype are now arrhythmic compared with those expressing p38Kb wt in a wild-type background (34% compared with 60%). Interestingly, although a larger proportion of flies now display robust free-running rhythms, the period length is extended even more than in flies that express p38K wt alone (Fig. 5, Table 3, and Suppl. Figs. S3 and S4, tau of 24.8 h for p38Kb wt alone and tau of 28.6 h with loss of per). Incidentally, loss of a single copy of per does not greatly affect the tau or arrhythmicity of the p38Kb asp phospho-mimic line. Furthermore, loss of per leads to an enhancement of both the p38Kb ala2 phospho-null (tau of 25.2 h and 7% arrhythmic to a tau of 27.6 h and 43% arrhythmic) and the p38Kb KD dominant negative backgrounds (tau of 25.5 h and 16% arrhythmic to a tau of 28.7 h and 52% arrhythmic, Fig. 5, Table 3, and Suppl. Figs. S3 and S4). Therefore, we tested whether p38Kb signaling may affect the cycling of phosphorylated PER. The PER protein is hyperphosphorylated by the kinases GSK3 (shaggy) and casein kinase II (CKII, double time); a recent study found that some PER phosphorylation sites are phosphorylated by an unknown kinase (Garbe et al., 2013). This phosphorylation state of PER dictates its subcellular localization, which is critical for PER function in regulating the clock. Therefore, we investigated the effect of altering p38Kb signaling on PER protein oscillations in phosphorylation. We find that control animals have robust oscillations in PER under both LD and DD as described by Edery et al. (1994). Although inhibition of p38Kb does not significantly alter the timing of these oscillations under DD conditions, it may affect the levels of PER during DD (Fig. 6) as recently observed by Dusik et al. (2014). Interestingly, we do not observe a significant increase in PER levels when p38Kb wt is overexpressed, suggesting that p38Kb-mediated PER phosphorylation is a tightly regulated process. However, the strong transheterozygous interaction between per and p38Kb lends further support to recent findings that p38Kb inhibition affects PER subcellular localization and phosphorylation and that p38Kb can phosphorylate PER in vitro (Dusik et al., 2014).
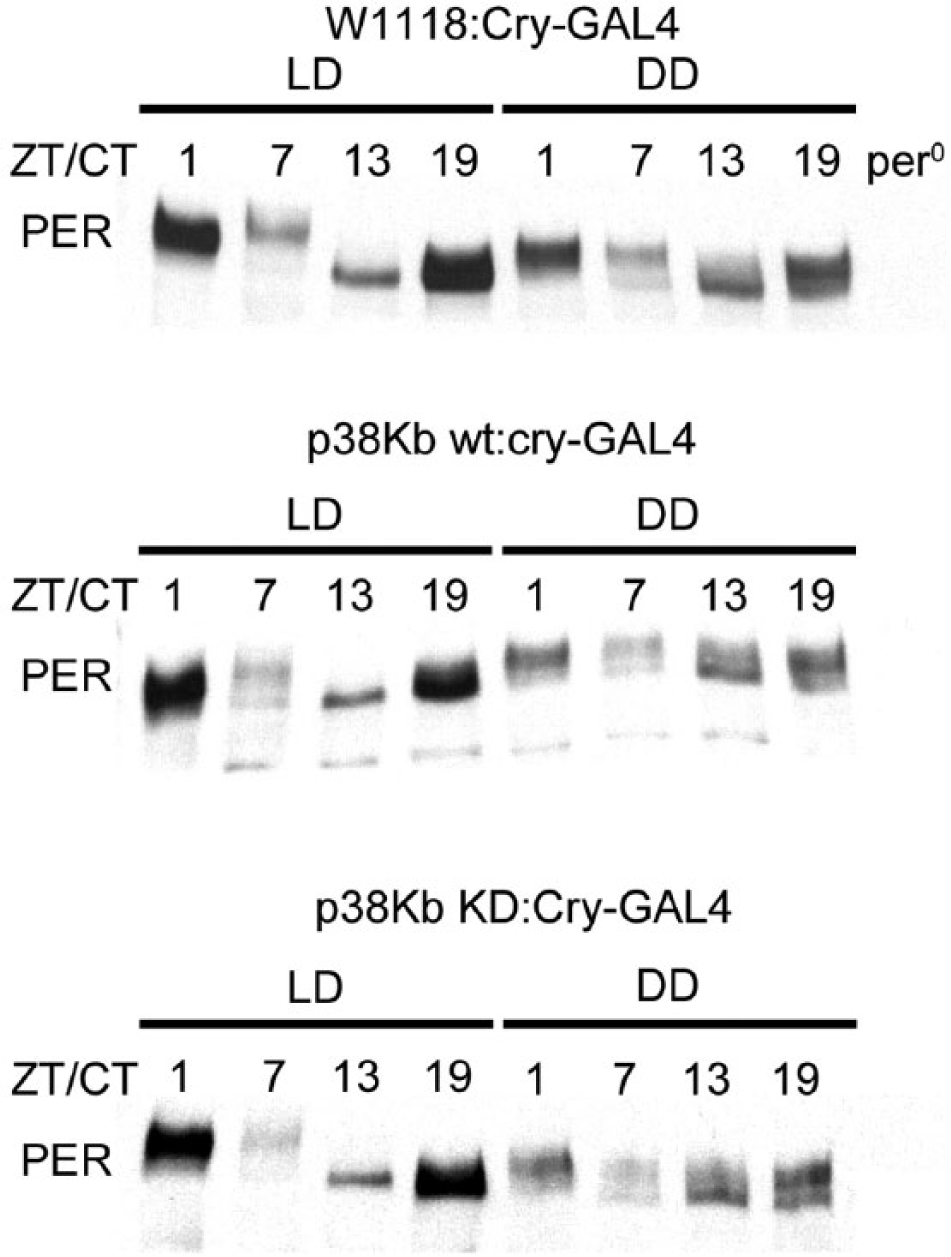
PER cycling under free-running conditions is not regulated by p38Kb. Animals were entrained in a 12-h:12-h light-dark cycle (LD) and collected on the second day of constant darkness (DD). Perturbations in p38Kb do not affect the cycling of PER hyperphosphorylation state throughout the 24-h clock. However, inhibition of p38Kb results in a decrease in overall PER levels.
To determine whether PER might be a target of p38Kb in vivo, we used the Cry-GAL4 to drive clock neuron expression of p38Kb KD, which is FLAG tagged. The protein product of the p38Kb KD mutant gene is thought to be able to bind to downstream targets but has reduced efficiency in subsequent release (Robinson et al., 1996), thus providing a unique opportunity to observe potentially transient interactions. We immunoprecipitated p38Kb KD from whole heads collected at ZT1, when PER levels are high, and assayed for binding to PER. Although p38Kb was recently shown to phosphorylate PER in vitro (Dusik et al., 2014), we were unable to detect PER binding to p38Kb KD in vivo. It may be that the p38Kb-PER interaction is too transient for us to detect even using the p38Kb KD. Another possibility is that p38Kb-mediated phosphorylation of PER is tightly regulated temporally and spatially. For example, p38Kb may only phosphorylate PER at night when PER is cytoplasmic rather than nuclear as it is at ZT1 (Curtin et al., 1995).
p38Kb Genetically Interacts with Mef2 to Regulate Circadian Rhythms
As p38 MAPKs have a number of downstream targets, p38Kb may be regulating other factors that give rise to circadian locomotor behaviors. We recently found that p38Kb acts through the transcription factor Mef2, a bona fide p38K target, to regulate aging and oxidative stress (Vrailas-Mortimer et al., 2011). Mef2 is transcriptionally regulated by the CLK/CYC complex (Sivachenko et al., 2013) and expressed in both the l-LNv and s-LNv neurons (Blanchard et al., 2010). Expression of a dominant negative Mef2 (Mef2 EnR) in which the transcriptional activation domain has been replaced with the Engrailed transcriptional repression domain results in arrhythmia, as reported previously (Fig. 7 and Table 4, Blanchard et al., 2010). We find that overexpression of p38Kb wt in the Mef2 EnR background rescues the arrhythmia phenotype from 84% to 53% arrhythmic (Fig. 7 and Table 4). Furthermore, p38Kb wt overexpression alone exhibits an increased tau, which is further magnified by inhibition of Mef2 (24.8 h and 26.6 h, respectively (Fig. 7 and Table 4), similar to what we observe when removing a single copy of per in the p38Kb wt overexpression background. These data suggest that p38Kb may regulate circadian rhythms through interactions with multiple downstream targets.
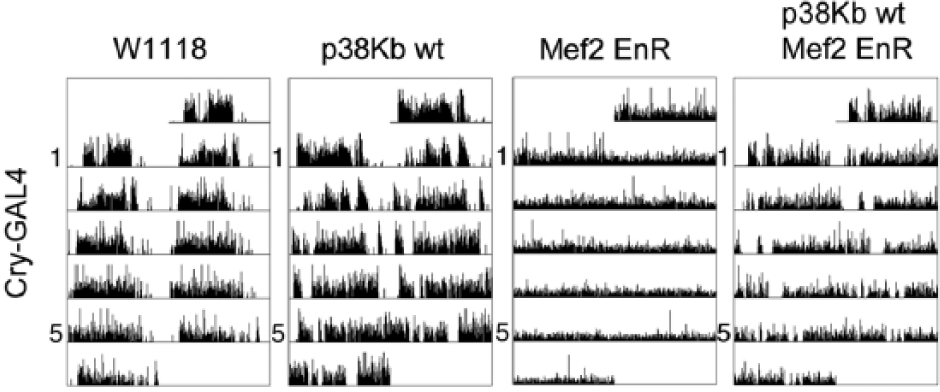
p38Kb genetically interacts with Mef2 to regulate circadian locomotor behaviors. Expression of dominant negative Mef2 (Mef2 EnR) driven by Cry-GAL4 results in arrhythmia, which is partially rescued by p38Kb wt overexpression.
p38Kb genetically interacts with Mef2.
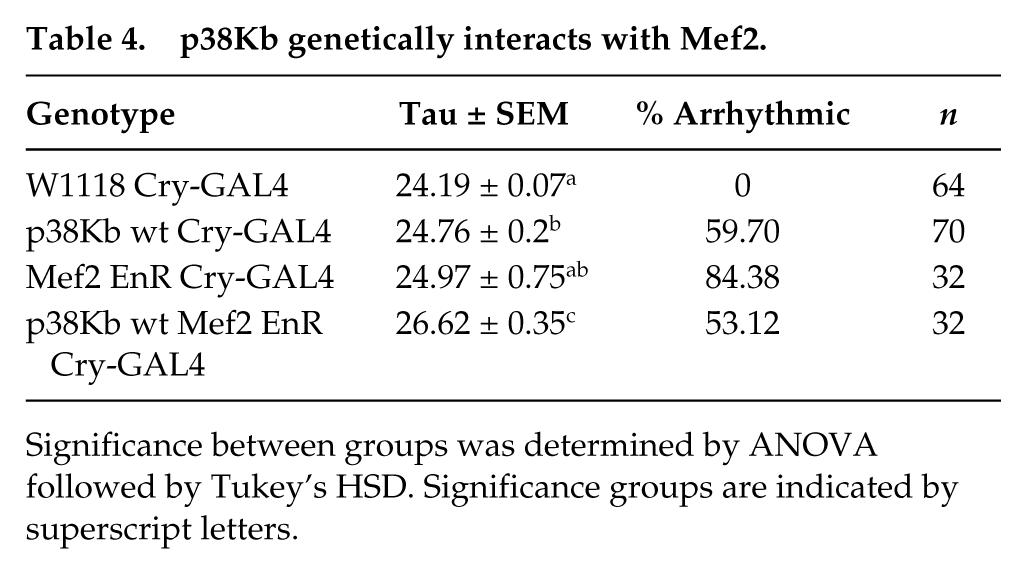
Significance between groups was determined by ANOVA followed by Tukey’s HSD. Significance groups are indicated by superscript letters.
p38Kb Signaling Affects MNK/Lk6 Circadian Cycling
In mammalian systems, another downstream target of p38K is the MAPK interacting kinase MNK/Lk6, which phosphorylates the AKT downstream target eIF4E. Upregulation of the AKT pathway leads to an increased period length in flies (Zheng and Sehgal, 2010), and Drosophila Lk6 has been shown to be transcriptionally regulated by CLOCK in whole heads (Abruzzi et al., 2011) and shows circadian oscillations in transcript expression (Dubruille et al., 2009). lk6 mutants are arrhythmic under constant light conditions, whereas overexpression of lk6 leads to an increased period length (Dubruille et al., 2009). Therefore, we tested whether p38Kb also regulates Lk6 expression in clock neurons. We found that Lk6 is expressed in the l-LNv clock neurons in the adult brain (where we observed expression of the p38Kb transcriptional reporter) and that levels of Lk6 seem to be aberrantly regulated upon p38Kb inhibition such that virtually no Lk6 is observed at CT13 whereas elevated levels of Lk6 are seen at CT19 compared with clock neurons in control brains (Fig. 8). Thus, p38K may be regulating circadian rhythms by influencing multiple downstream targets.
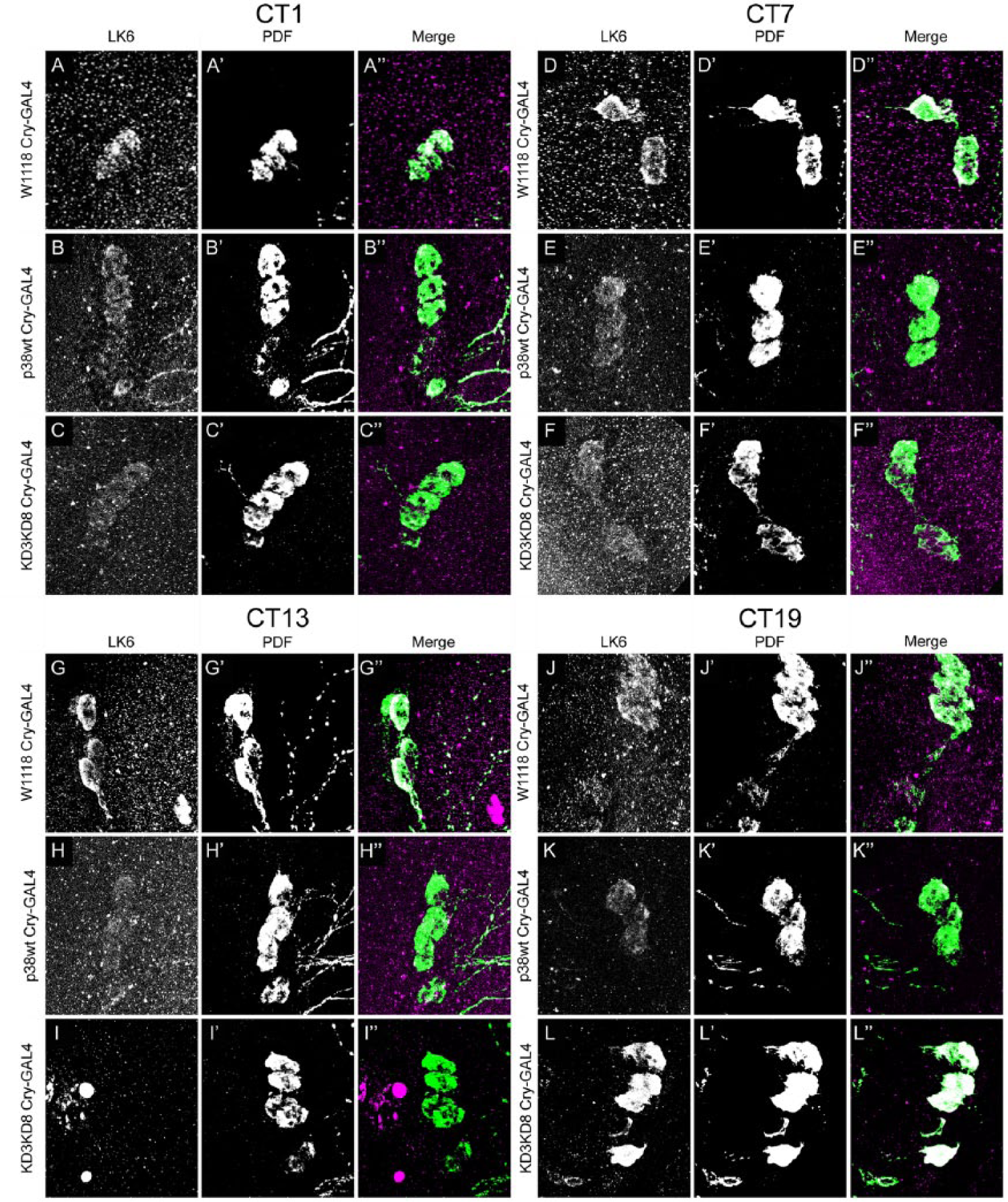
p38Kb regulates Lk6 expression in clock neurons throughout the clock. Animals were entrained in a 12-h:12-h light-dark (LD) cycle and then placed in constant darkness (DD) and collected during the second day of DD. Lk6 is expressed in PDF positive l-LNv clock neurons (A, D, G, and J). Lk6 expression is decreased at CT13 when p38Kb is overexpressed (compare H with G). Inhibition of p38Kb results in decreased Lk6 expression at CT13 (compare I with G) but increased expression at CT19 (compare L with J). Merged images are LK6 (magenta) and PDF (green), and white shows colocalization.
Discussion
The ability of an organism to properly respond to its environment is, in part, influenced by circadian rhythms (Bartok et al., 2013; Kondratov et al., 2006; Krishnan et al., 2008; Zheng et al., 2007), suggesting that stress response pathways may play an important role in maintaining these rhythms. Interestingly, manipulating aging genes in clock neurons, which are tightly linked to stress response, negatively affects circadian rhythms (Zheng et al., 2007). Therefore, other genes linked to stress and aging, such as the stress-activated protein kinase p38K, may also play roles in regulating circadian rhythms. Our data suggest that p38Kb contributes to maintaining a proper circadian clock with either inhibition or promiscuous activation of p38Kb leading to aberrant rhythms. Flies have two p38K genes, p38Ka and p38Kb, that have differential levels of expression in the fly brain, with p38Kb as the predominant species (Vrailas-Mortimer et al., 2011). Furthermore, we find that p38Kb is expressed in clock neurons and that loss of either gene alone at a young age does not influence circadian locomotor behavior. However, we have previously found that p38Kb mutants have many age-dependent phenotypes (Vrailas-Mortimer et al., 2011), and consistent with these observations, we find that aged p38Kb mutant animals have a minor but significant period lengthening in free-running conditions. In younger animals, p38Kb-dependent phenotypes require much stronger knockdown, perhaps due to the ubiquitous and high level of p38Kb expression in the brain. Indeed, we notice a large percentage of arrhythmic flies when both p38Ka and p38Kb levels are reduced (p38K DKO). Additionally, significant period lengthening is observed when p38Kb is inhibited in clock neurons using the Cry-GAL4 or Pdf 0.5-GAL4 driver lines in conjunction with either a phospho-null (p38Kb ala2) or a kinase-dead version of p38Kb (p38Kb KD). These results also confirm that p38Kb perturbation in clock neurons is sufficient to precipitate circadian phenotypes, which was recently observed by Dusik et al. (2014). Interestingly, Dusik et al. (2014) also observed that knockdown of p38Ka results in period lengthening. MAPKs are known to homodimerize, which is important for cytoplasmic functions as well as nuclear translocation (Khokhlatchev et al., 1998). The p38Ka and p38Kb amino acid sequences are 78% identical and have 94% sequence similarity and have been found to co-Immunoprecipitate (co-IP) with each other (Belozerov et al., 2014; Guruharsha et al., 2011). Therefore, p38Ka and p38Kb likely act as both homodimers and heterodimers, which may have specific functions. The phospho-null or kinase dead forms of p38Kb may inhibit p38Kb homodimer and heterodimer functions but may also block the formation of p38Ka homodimers, which normally would be able to compensate for the lack of p38Kb homodimers and heterodimers. RNAi knockdown of either p38Kb or p38Ka also leads to period lengthening (Dusik et al., 2014), which is not too surprising as the mRNA coding sequences of p38Kb and p38Ka are 73% identical, and these RNAi lines target regions in common for p38Ka and p38Kb. Thus, p38Ka and p38Kb may have compensatory roles in regulating circadian rhythm, and inhibition of both proteins is necessary to elicit a phenotype.
Activation of p38Kb in clock neurons, achieved through the expression of either wild-type p38Kb or a phospho-mimic p38Kb (p38Kb asp), has the same effect as p38Kb inhibition in extending the length of the circadian period. This seemingly contradicts the simple expectation that p38Kb overexpression might shorten period length. Therefore, we cannot assume a simplistic model of p38Kb in the regulation of circadian rhythms. One idea for why bidirectional changes in p38Kb produce the same phenotype (period lengthening) is that in both cases, oscillations in either p38Kb expression or activity are dampened. Another possibility is that p38Kb is required for the regulation of a downstream target that is under the control of a cycling mechanism.
As p38Kb is a regulator of the stress response and loss of p38Kb results in increased oxidative stress and reduced lifespan (Vrailas-Mortimer et al., 2011), it is possible that p38Kb perturbations adversely affect the health of clock neurons. However, the number of PDF positive neurons is not reduced following p38Kb inhibition (data not shown). Since we observe p38Kb-dependent phenotypes only in complete darkness, we hypothesized that p38Kb may affect the core clock machinery, in particular PER, since per mutants display similar behavioral phenotypes in free-running conditions. Indeed, we find that p38Kb genetically interacts with per to regulate period length and arrhythmicity. Furthermore, we find that inhibition of p38Kb leads to an overall decrease in PER protein levels in free-running conditions as recently described by Dusik et al. (2014). PER phosphorylation state also oscillates in a circadian manner, with PER having less phosphorylation during early night and becoming increasingly hyperphosphorylated throughout the rest of the night and into the day (Edery et al., 1994). Many of these PER phosphorylation events have been mapped to the kinases GSK3 and CKII. However, Garbe et al. (2013) identified additional phosphorylation sites that have not been experimentally linked to a particular kinase. In addition, Dusik et al. (2014) recently reported that p38Kb can phosphorylate PER in vitro. Although we found that p38Kb genetically interacts with per to regulate both tau and arrhythmicity, we were unable to detect significant differences in PER hyperphosphorylation levels when p38Kb wt is overexpressed, suggesting that p38Kb may be able to phosphorylate PER only under certain conditions such as when PER is cytoplasmic or has reached a specific phosphorylation state.
Although p38Kb may be regulating PER, p38 MAPKs have a variety of downstream targets that may also contribute to maintaining circadian rhythms. One such downstream target is Mef2, which, like p38Kb, results in period length extension and increased arrhythmia when overexpressed or inhibited (Blanchard et al., 2010). We find that p38Kb genetically interacts with Mef2 in regulating circadian locomotor behavior, with overexpression of p38Kb wt rescuing the arrhythmia observed by Mef2 inhibition. Although Mef2 has been shown to cycle in the s-LNv neurons (Blanchard et al., 2010), the Mef2 phosphorylation state in these cells is not known. In addition, Mef2 is also expressed in the l-LNv neurons, although Mef2 cycling in these neurons has yet to be examined. One possibility is that manipulating p38Kb in the s-LNvs interferes with p38Ka activity in these neurons, resulting in dysregulation of Mef2, although Mef2 has yet to be shown as a downstream target of p38Ka. Alternatively, p38Kb may regulate Mef2 in the l-LNv neurons, which are hypothesized to integrate multiple circadian inputs and transmit this information to multiple outputs including the s-LNv neurons (reviewed in Helfrich-Forster, 2005). As the relationship between the l-LNv and s-LNv neurons is not fully understood, further insights may be gained by observing how manipulating signaling cascades in the l-LNv neurons influences the s-LNv neurons.
Another downstream target of p38K is the MAPK interacting kinase MNK/Lk6, the transcription of which oscillates in a circadian fashion and is regulated by CLK/CYC (Claridge-Chang et al., 2001; Dubruille et al., 2009). We find that Lk6 protein is indeed expressed in clock neurons and upon p38Kb inhibition shows aberrant expression compared with clock neurons in control brains. This indicates that p38Kb perturbations can affect MNK/Lk6 function in clock neurons, although it remains to be seen whether this is a direct effect or occurs indirectly through, for instance, the CLK/CYC complex. Overall, these results suggest that p38Kb may be regulating multiple signals that contribute to circadian rhythm locomotor behavior. If p38Kb is functioning as a nodal point to integrate several upstream signaling inputs as well as relaying signaling events to a large number of downstream effectors in clock neurons, then the level of synchrony that is required to maintain a normal circadian rhythm must be exquisite. Studying these multiple p38Kb-dependent signaling pathways and their oscillations simultaneously is a prerequisite for understanding the complexity and plasticity in this system.
Footnotes
Acknowledgements
The authors thank Justin Blau and Amita Sehgal for useful discussions and the gift of Pdf 0.5-GAL4 and UAS-Mef2 EnR and anti-PER antibodies, respectively. The authors also thank Verena Dusik and Charlotte Helfrich-Forster for sharing their unpublished results and useful comments. This study was supported by grants from the Brain and Behavior Research Foundation (formerly the National Alliance for Research on Schizophrenia and Depression) and by National Institutes of Health Grants 1R03DA027979 and 5R21MH091520 to S.S., a Center for Behavioral Neuroscience NSF postdoctoral fellowship, a PD-CERC T32 ES012870 fellowship, and start-up funds from the University of Denver to A.D.V.M. All research materials are available upon request.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.