
Abstract
Endogenous 24-h rhythms in physiology are driven by a network of circadian clocks located in most tissues. The molecular clock mechanism is based on feedback loops involving clock genes and their protein products. Posttranslational modifications, including ubiquitination, are important for regulating the clock feedback mechanism. Recently, we showed that the deubiquitinating enzyme ubiquitin-specific peptidase 2 (USP2) associates with clock proteins and deubiquitinates PERIOD1 (PER1) but does not affect its overall stability. Mice devoid of USP2 display defects in clock function. Here, we show that USP2 regulates nucleocytoplasmic shuttling and nuclear retention of PER1 and its repressive role on the clock transcription factors CLOCK and BMAL1. The rhythm of nuclear entry of PER1 in Usp2 knockout mouse embryonic fibroblasts (MEFs) was advanced but with reduced nuclear accumulation of PER1. Although Per1 mRNA expression rhythm remained intact in the Usp2 KO MEFs, the expression profiles of other core clock genes were altered. This was also true for the expression of clock-controlled genes (e.g., Dbp, Tef, Hlf, E4bp4). A similar phase advance of PER1 nuclear localization rhythm and alteration of clock gene expression profiles were also observed in livers of Usp2 KO mice. Taken together, our results demonstrate a novel function of USP2 in the molecular clock in which it regulates PER1 function by gating its nuclear entry and accumulation.
Daily oscillations exist for most behavioral and physiological functions of mammals. Such rhythms are generated and maintained by a network of endogenous circadian clocks. The central clock in mammals, located in the suprachiasmatic nucleus (SCN) of the hypothalamus, coordinates with varying degrees of control the clocks found in other brain regions and most peripheral tissues (Okamura, 2007; Cuninkova and Brown, 2008). Two major features characterize clock function: the self-sustainability of the system as revealed by persistent circadian rhythm generation in the absence of environmental cues and its capacity to respond to cues (such as light, temperature, feeding), which enables synchrony with environmental cycles and within the organism. Central and peripheral clocks influence a vast number of processes, including development, life span, epigenetic modifications, and metabolism (Yu and Weaver, 2011; Feng and Lazar, 2012; Liu and Chu, 2013). Correspondingly, disturbances or misalignment of the circadian clock system, such as those occurring in shift workers, have been associated with a wide variety of disorders such as cancer, metabolic syndrome, obesity, diabetes, and cardiovascular diseases (Morikawa et al., 2005; Kivimaki et al., 2006; Gery and Koeffler, 2010; Yu and Weaver, 2011; Bass, 2012; Liu and Chu, 2013).
The molecular mechanism underlying these circadian rhythms is based on transcriptional/translational feedback loops involving clock genes and proteins, as well as posttranslational modifications of the clock proteins (Duguay and Cermakian, 2009). In the core loop, BMAL1 and CLOCK proteins form a heterodimer that drives the transcription of genes, including the Period (Per) 1 and 2 and Cryptochrome (Cry) 1 and 2 genes through E-box elements located in their promoter regions. The PER and CRY proteins accumulate in the cytoplasm, where they are subjected to posttranslational modifications and subsequently enter the nucleus to repress the activity of CLOCK/BMAL1 and thus to suppress their own transcription. Furthermore, CLOCK/BMAL1 also stimulate the expression of other transcription factors such as the nuclear receptor REV-ERBα and the PAR bZip factors DBP (albumin D-box binding protein), TEF (thyrotroph embryonic factor), and HLF (hepatic leukemia factor). REV-ERBα suppresses the transcription of Bmal1 as well as that of E4bp4 (adenovirus E4 binding protein 4, also called Nfil3). The net effect of the molecular clock is a rhythmic change in the activity of different transcription factors and thereby the rhythmic expression of thousands of output genes, termed clock-controlled genes. These clock-controlled genes cycle in various tissues and cell types and participate in numerous biological processes, such as cell cycle regulation and metabolism (Sahar and Sassone-Corsi, 2009).
The timing of both the entry and the residence time of core clock proteins in the nucleus is critical for maintaining the correct pace and functional integrity of the clock mechanism. Immunohistochemical studies on SCN neurons have revealed that CRY1, CRY2, PER1, and PER2 proteins undergo near-synchronous circadian patterns of nuclear abundance (Field et al., 2000). These clock proteins, as well as BMAL1 and REV-ERBα, contain nuclear entry and export signals that are modulated to precisely regulate clock protein nucleocytoplasmic shuttling (Miyazaki et al., 2001; Yagita et al., 2002; Chopin-Delannoy et al., 2003; Tamanini et al., 2005). This dynamic intracellular movement influences function by determining interacting partners and protein turnover in a time-dependent fashion (Kwon et al., 2006). The time-dependent regulation of clock protein “shuttling” is still poorly understood, but it is believed to be modulated by posttranslational modification involving phosphorylation, such as casein kinase Iϵ-mediated phosphorylation of PER1 (Takano et al., 2004; Virshup et al., 2007), ubiquitination, and subsequent proteasomal degradation.
The ubiquitin-proteasome system plays an important role in the daily oscillations in abundance and activity of the clock proteins. In this system, ubiquitin is conjugated to target proteins through the sequential action of 3 types of enzymes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin protein ligases (E3) (Glickman and Ciechanover, 2002). E3s play the critical role of recognizing the substrate for ubiquitination. Formation of polyubiquitin chains on substrates, particularly Lys48-linked chains where the C-terminus of the distal ubiquitin moiety is linked to lysine 48 of the more proximal ubiquitin moiety, targets the substrate for recognition and degradation by the proteasome. Mammalian E3s can regulate the stabilities of PER and CRY proteins. For example, silencing or mutation of the E3 substrate receptors β-TRCP1/2 results in stabilization of PER proteins and altered circadian rhythms in cultured cells (Grima et al., 2002; Shirogane et al., 2005; Reischl et al., 2007; Ohsaki et al., 2008). Another pair of E3 substrate binding proteins, FBXL3 and FBXL21, act on CRY proteins (Busino et al., 2007; Siepka et al., 2007; Dardente et al., 2008; Hirano et al., 2013; Yoo et al., 2013). Notably, despite the sequence similarity of these 2 enzymes, their roles in the clock are distinct. FBXL3 targets CRY proteins for proteasomal degradation, and loss-of-function mutation results in stabilization of CRY proteins and lengthening of the free-running period (Busino et al., 2007; Siepka et al., 2007;Godinho et al., 2007). In contrast, FBXL21 stabilizes CRY proteins and favors their retention in the cytoplasm, and its absence causes a short-period phenotype in mice and cells (Hirano et al., 2013; Yoo et al., 2013). Finally, the E3 ligases Huwe1 (Arf-bp1) and Pam are involved in regulating the degradation of REV-ERBα (Yin et al., 2010).
As occurs for other covalent modifications such as protein methylation and phosphorylation, ubiquitination is reversible. Deubiquitination is catalyzed by approximately 100 deubiquitinating enzymes (DUBs), belonging to 5 families (Komander et al., 2009; Sowa et al., 2009). These enzymes remove ubiquitin from target proteins and can thereby reverse the effects of ubiquitination. Thus, it is possible that DUBs regulate clock proteins through removal of degradation signals attached by β-TRCP1/2, FBXL3/21, and other ligases. We previously identified the DUB ubiquitin-specific peptidase 2 (USP2) from rat testis (formerly, UBP-testis) (Lin et al., 2000; Lin et al., 2001). Analysis of circadian microarray data from multiple tissues reveals that the gene encoding USP2 has a rhythmic circadian expression pattern (Storch et al., 2002; Yan et al., 2008), a feature shared only by a very limited number of genes, including core clock components (Yan et al., 2008). Thus, we set out to test the role of USP2 in circadian clock control using gene targeting and cellular studies. We previously found that USP2 acts as an integral component of the circadian clock with Usp2–/– mice having abnormalities in both speed of the endogenous clock as well as its entrainment by photic cues (Yang et al., 2012). At a molecular level, we demonstrated that PER1 is highly ubiquitinated in cells and is a substrate of USP2, but deubiquitination of PER1 does not affect overall stability of the protein (Yang et al., 2012). We therefore explored whether USP2 might regulate PER1 in a nondegradative manner. To this end, we tested whether USP2 modulates the nucleocytoplasmic shuttling of PER1 protein and thereby its effects on core clock mechanisms.
Materials And Methods
Maintenance of Usp2 KO Mice and Mouse Liver Sample Collection
Mice lacking functional USP2 were generated by gene targeting as described previously (Bedard et al., 2011). Wild-type (WT) and Usp2 KO female mice (mostly 12-13 [range, 9-17] months of age) were generated by breeding of heterozygous mice whose genetic backgrounds were 98.4% C57BL/6 (5 backcrosses with C57BL/6, starting from 50:50 C57BL/6:129Sv mice). These mice were entrained to a 12-h light, 12-h dark (LD 12:12) cycle. Usp2 knockout (KO) mice and WT littermates were sacrificed every 3 h over 24 h starting at zeitgeber time 1 (ZT1, 4, 7, 10, 13, 16, 19, 22, where ZT0 is the time of lights-on and ZT12 is the time of lights-off). For nighttime points, mice were sacrificed under dim red light. Liver samples were frozen in liquid nitrogen or prechilled isopentane for RNA and immunostaining analyses, respectively. Procedures involving animals were carried out in accordance with guidelines of the Canadian Council on Animal Care and approved by the Animal Care Committees of McGill University and Douglas Mental Health University Institute.
Reagents, Plasmids, and cDNA Constructs
Cell culture media and Lipofectamine 2000 were from Life Technologies (Burlington, ON, Canada); protease inhibitors (Mini Cocktail) were from Roche (Mississauga, Ontario, Canada). Antibodies used in the study are showed in the Supplementary Table S1.
Plasmids expressing Flag-tagged CLOCK (pSG5-mCLOCK-Flag), Myc-tagged BMAL1 (pCS-MTK-mBMAL1-Myc), V5-tagged PER1 (pCDNA3.1-mPER1-V5-His6), HA-tagged CRY1 (pCDNA3.1-mCRY1-HA), luciferase reporter under control of the Per1 promoter (pGL3-basic-mPer1 promoter [−1803 to +40]-luciferase), C-terminal Flag- or HA-tagged rat USP2a (AF202453, pRK5-USP2a-Flag, or HA), USP2b (AF202454, pRK5-USP2b-Flag, or HA), and the corresponding catalytic inactive USP2 mutants (USP2a C288A, and USP2b C67A) were described previously (Lin et al., 2000; Lin et al., 2001; Travnickova-Bendova et al., 2002; Yang et al., 2012), and green fluorescent protein (GFP)–expressing plasmid (pEGFP-C3) was from Life Technologies.
Cell Culture, Transfections, and Immunoblotting
HEK293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. At 80% to 90% confluence, cells were transfected with the indicated plasmids or the corresponding empty vectors to a total amount of 0.1 µg DNA/cm2 surface area of culture vessel using Lipofectamine 2000 following the manufacturer’s protocol. After 48 h, cells were rinsed twice in cold phosphate-buffered saline (PBS) and lysed in TTE (20 mM Tris-Cl [pH 8.0], 1% Triton X-100, 5 mM EDTA, 150 mM NaCl, 5 mM NaF, 5 mM Na pyrophosphate, 1 mM Na3VO4, 1 mM PMSF). Proteins, quantified by the Micro BCA assay kit (Pierce, Rockford, IL), were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes. The membranes were incubated with the indicated primary antibodies, followed by goat anti–mouse/rabbit secondary antibody conjugated to horseradish peroxidase (HRP), and finally with chemiluminescent substrate (ECL, Amersham, Piscataway, NJ or PerkinElmer, Waltham, MA), and the signals were detected with a cooled digital camera (Bio-Rad Molecular Imager VersaDoc MP 4000 System; Bio-Rad, Hercules, CA).
Synchronization of Mouse Embryonic Fibroblasts With Serum Shock
WT and Usp2 KO mouse embryonic fibroblasts (MEFs), generated as previously described (Yang et al., 2012), were grown in DMEM with 10% FBS, 2 mM L-glutamine, and 1% penicillin-streptomycin. They were synchronized as described previously (Balsalobre et al., 1998), with minor modifications. Briefly, cells were plated in 6-well plates at 1 × 105 cells/well and in 35-mm dishes covered with circle coverslips at 0.8 × 106 cells/dish and then grown to confluence. After an overnight serum starvation (15 h), cells were exposed to 50% horse serum for 2 h, followed by replacement with serum-free DMEM supplemented with 2 mM L-glutamine and 1% penicillin-streptomycin. At indicated times (the starting point when 50% horse serum is added was set as T0), WT and Usp2 KO MEFs cells were processed as follows. For immunostaining analysis, cells grown on coverslips were washed 3 times with PBS, 1 mM CaCl2, and 0.5 mM MgCl2; fixed and kept in 4% paraformaldehyde in PBS at 4 °C; and used for immunostaining within 1 week. For RNA analysis, cells from each well of 6-well plates were lysed in 1 mL TRIzol (Life Technologies) and the RNA was isolated as per the manufacturer’s protocol.
Immunofluorescence Staining and Confocal Microscopy
To determine the intracellular distribution of PER1 and its modulation by USP2, HEK293 cells at 95% confluence were transfected with plasmids expressing PER1-V5 and GFP (20:1 w/w ratio), with or without USP2b-Flag. The next day, the cells were replated at 1:5 dilution to reduce cell densities and grown on cover slips for better observation by microscopy. Forty-eight hours after transfection, the cells were fixed in 4% paraformaldehyde/PBS, permeabilized in 0.5% Triton X-100/PBS, blocked with 0.25% bovine serum albumin (BSA)/PBS, and then incubated for 1 h without (control) or with anti-V5 antibody, followed by an Alexa Fluor 568 goat anti–mouse antibody and then brief staining with 25 µg/mL 4′,6-diamidino-2-phenylindole-dihydrochloride (DAPI) to detect the nuclei. The paired control and experimental coverslips were mounted side by side onto a single slide. Confocal images were obtained from a Carl Zeiss Laser Scanning confocal microscope (LSM510; Carl Zeiss [Toronto, Ontario, Canada]), and the intracellular localization of PER1 was determined based on the distribution of fluorescent signals present in 40 to 90 GFP-positive cells.
To determine the intracellular distribution of endogenous PER1 and its modulation by USP2, WT and Usp2 KO MEFs grown on coverslips and synchronized by serum shock were subjected to a similar immunostaining procedure as the transfected HEK293 cells. The coverslips, however, were incubated overnight without (control) or with a rabbit anti-PER1 antiserum, followed by an Alexa Fluor 488 goat anti–rabbit antibody for 1 h and then brief staining with DAPI.
To determine the circadian expression, rhythm, and intracellular shuttling of endogenous PER1 and its modulation by USP2 in vivo, liver sections obtained at different ZT points from paired WT and Usp2 KO mice were immunostained for PER1 and USP2, as described previously (Fahrenkrug et al., 2006). Briefly, frozen sections (9 µm) were fixed in 4% paraformaldehyde (PFA)/PBS for 20 min and incubated overnight without (control) or with rabbit anti-PER1 antiserum, or rabbit anti-USP2 antibody, followed by incubation with Alexa Fluor 488 goat anti–rabbit and Alexa Fluor 568 goat anti–rabbit antibody for 1 h, respectively. Photomicrographs were obtained with an Olympus IX70 confocal microscope equipped with Fluoview (version 2.1.39; Olympus, Tokyo, Japan).
Luciferase Reporter Assays
HEK293 cells were transfected in 24-well plates with DNA mix containing luciferase gene reporter (25 ng pGL3-basic-mPer1), with or without the indicated expression constructs (200 ng pSG5-mCLOCK, 200 ng pCS2-MTK-mBMAL1, 50 or 200 ng pRK5-mUSP2, 50 ng PER1, 50 ng CRY1), adjusted with the corresponding empty vectors to a total of 625 ng to study the effect of USP2, PER1, and CRY1 on CLOCK/BMAL1 activity. The gene reporter assays were then performed and normalized as previously described (Dardente et al., 2007). Briefly, cells were rinsed twice with ice-cold PBS and exposed to 150 µL lysis buffer. After a short centrifugation step to pellet cell debris, the luciferase assay was carried out in luciferase buffer with 10 µL of cell lysate using the Orion II Microplate Luminometer (Berthold Detection Systems, Oak Ridge, TN). Data represent fold induction over the appropriate control condition. The graphs show data pooled from 3 separate experiments, each done in triplicate, which all showed very similar results. One-way analysis of variance (ANOVA) followed by post hoc tests (Bonferroni posttests) was used to compare conditions.
Quantitative PCR
RNA from liver samples was prepared by the guanidum isothiocyanate (Life Technologies) method (Ausabel et al., 1993), and RNA from cultured cells was prepared using Trizol reagent according to the manufacturer’s protocol. The purified RNA (1 µg) was used for cDNA synthesis using a reverse transcription kit (Life Technologies) following the manufacturer’s protocol. Quantitative PCR was performed on a ViiA 7 Real-Time PCR System (Life Technologies) using Power SYBR Green Master Mix reagent or TaqMan Universal Master Mix II reagent (Life Technologies) and 100 nM of the TaqMan probes or primer pairs for SYBR Green methods. Primer sequences for quantitative PCR are described in Supplementary Table S2. Primer efficiencies (for SYBR Green) were verified using serial dilutions of cDNA under the following conditions: 95 °C for 10 min, 40 cycles of 95 °C for 15 s, 50 °C for 30 s, and 72 °C for 30 s, followed by a melt curve stage at 95 °C for 15 s and then 60 °C for 1 min. Probes for TaqMan were used under the following conditions: 50 °C for 2 min, 95 °C for 10 min, and then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Values were normalized to β-Actin or Gapdh and then calibrated to WT time = 0 data for synchronized MEFs or to WT ZT1 data for mouse liver samples using the ΔΔCt method (Livak and Schmittgen, 2001). For cultured cell studies, data were pooled from 3 separate experiments, each carried out in triplicate. For the animal studies, there were 3 to 7 WT or KO mice sacrificed at each time point. Data were analyzed by 2-way ANOVA for time and genotype, followed by post hoc tests (Bonferroni posttests).
Results
USP2 Regulates the Intracellular Distribution of PER1
To test whether USP2 regulates the intracellular distribution of PER1 between the cytoplasm and the nucleus, we expressed PER1 in HEK293 cells, with or without USP2b. USP2 is expressed as 2 isoforms, USP2a and 2b, which share an identical core catalytic domain but distinct N-terminal extensions. The USP2b isoform has a more robust circadian rhythm of expression than the USP2a isoform (Kita et al., 2002; Storch et al., 2002; Oishi et al., 2005; McCarthy et al., 2007). Confocal microscopy showed that in most transfected cells, PER1 was present in the cytoplasm or in both cytoplasm and nucleus. Coexpressing USP2b led to a significant redistribution of PER1 to the nucleus with fewer cells expressing cytoplasmic PER1 only (Fig. 1A,B). Similar observations were made for USP2a (data not shown).
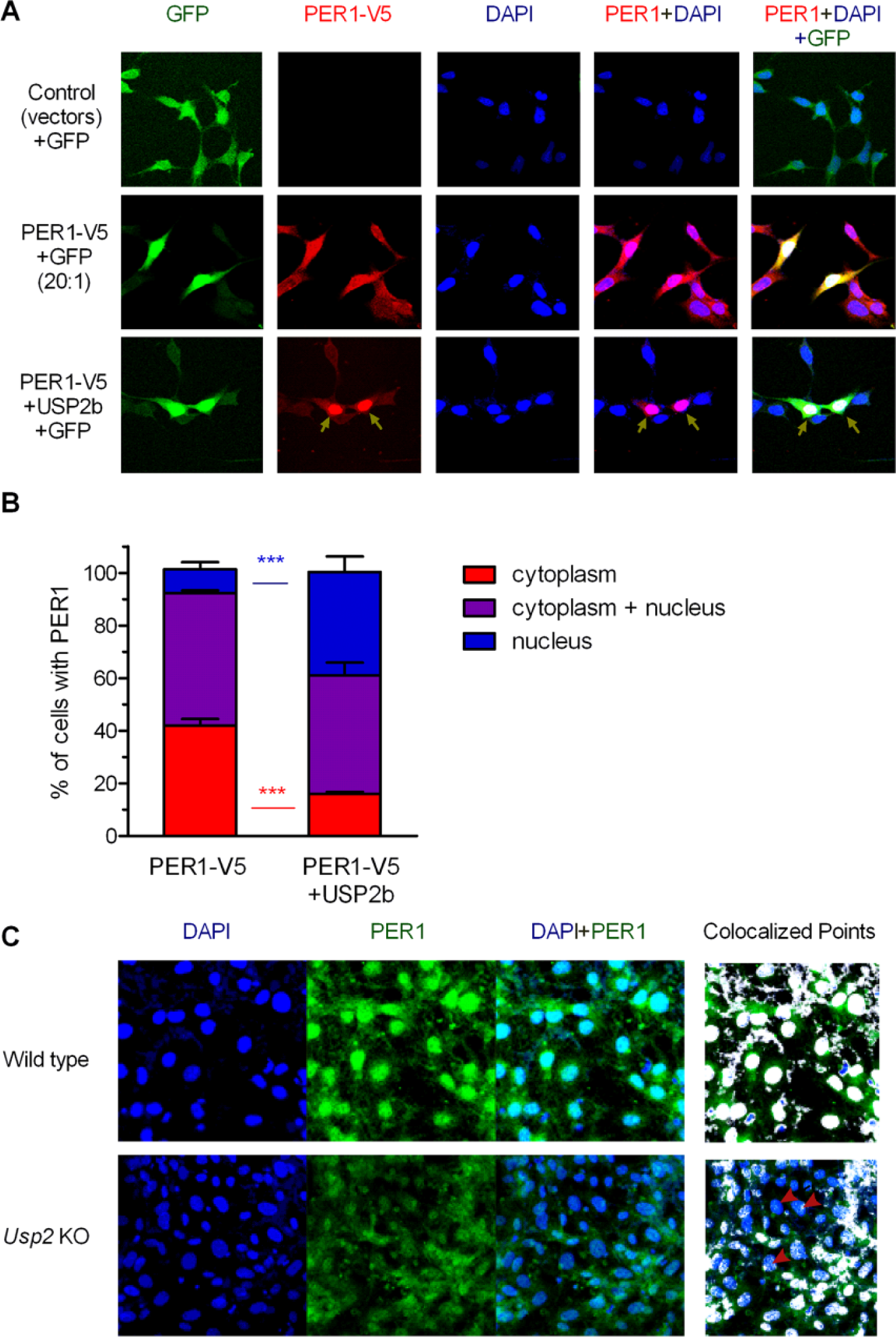
USP2 regulates the nuclear localization of PER1. (A) PER1-V5 was expressed alone or with USP2b (2:1 molar ratio of USP2b:PER1-V5 plasmids) in HEK293 cells. Green fluorescent protein (GFP) was coexpressed with PER1-V5 (1:10 molar ratio of the GFP:PER1-V5 plasmids) to identify transfected cells. PER1-V5 was immunostained with anti-V5 primary antibody and Alexa Fluor 568–conjugated secondary antibody (red). Cell nucleus was stained with DAPI (blue). Arrowheads indicate cells with predominantly nuclear localization. (B) The localization of PER1 was classified as predominantly cytoplasmic, predominantly nuclear, or both cytoplasmic and nuclear compartments based on the distribution of fluorescent signals present in 40 to 90 GFP-positive cells from 3 experiments. USP2b expression resulted in a significantly greater percentage of cells with predominantly nuclear localization of PER1 and a concomitant lower percentage of cells with cytoplasmic localization (shown are means ± SEM, p < 0.001, 2-way analysis of variance with Bonferroni posttest analyses). (C) Wild-type (WT) and Usp2 knockout (KO) mouse embryonic fibroblasts (MEFs) were serum starved overnight and then stained with an anti-PER1 antiserum followed by an Alexa Fluor 488 anti–rabbit IgG antibody (green) and with the nuclear stain DAPI (blue) (left panel). The PER1 and DAPI signals were analyzed using National Institutes of Health ImageJ software with colocalization plug-ins to identify nuclear localized PER1 signals, shown as white points (right panel). Shown are representative images of 3 independent experiments with similar results. Red arrowheads indicate examples of cells with decreased localization of PER1 in the nucleus.
We tested whether this was also true with endogenous PER1 by comparing its localization in WT and Usp2 KO MEFs (Fig. 1C). In WT MEFs, PER1 was found predominantly in the nucleus. In contrast, PER1 was detected more evenly in both cytoplasm and nucleus of the Usp2 KO MEFs. Taken together, these studies indicate that USP2 promotes nuclear entry or nuclear retention of the endogenous PER1.
USP2 Regulates PER1-Mediated Repression of CLOCK-BMAL1 Transcriptional Activity
Since USP2 promoted nuclear localization of PER1, we tested the effects of USP2 on PER1-mediated repression of CLOCK-BMAL1 transcriptional activity. This was achieved using a reporter in which luciferase expression is driven by a Per1 promoter fragment that contains the critical E-box elements. Expression of either isoform of USP2 reduced the transcriptional activity of CLOCK/BMAL1 in a dose-dependent manner (Fig. 2A). This was not observed with the catalytically inactive USP2-CA mutant, indicating that the enzymatic activity of USP2 was required for the effect. Furthermore, when transfected at levels that did not significantly alter CLOCK/BMAL1 activity (see Fig. 2A, second and fifth bars), both isoforms of USP2 acted additively with PER1 and CRY1 to attenuate CLOCK/BMAL1 transcriptional activity (Fig. 2B). Again, enzymatic activity of USP2 was required to obtain this effect. These results are unlikely to be due to a downregulation of CLOCK or BMAL1 since the levels of the tagged CLOCK and BMAL1 were not reduced in the presence of USP2 (Fig. 2C). Thus, USP2 appears to potentiate PER/CRY-mediated inhibition of CLOCK/BMAL1 activity by increasing the entrance or retention in the nucleus of the repressor proteins.
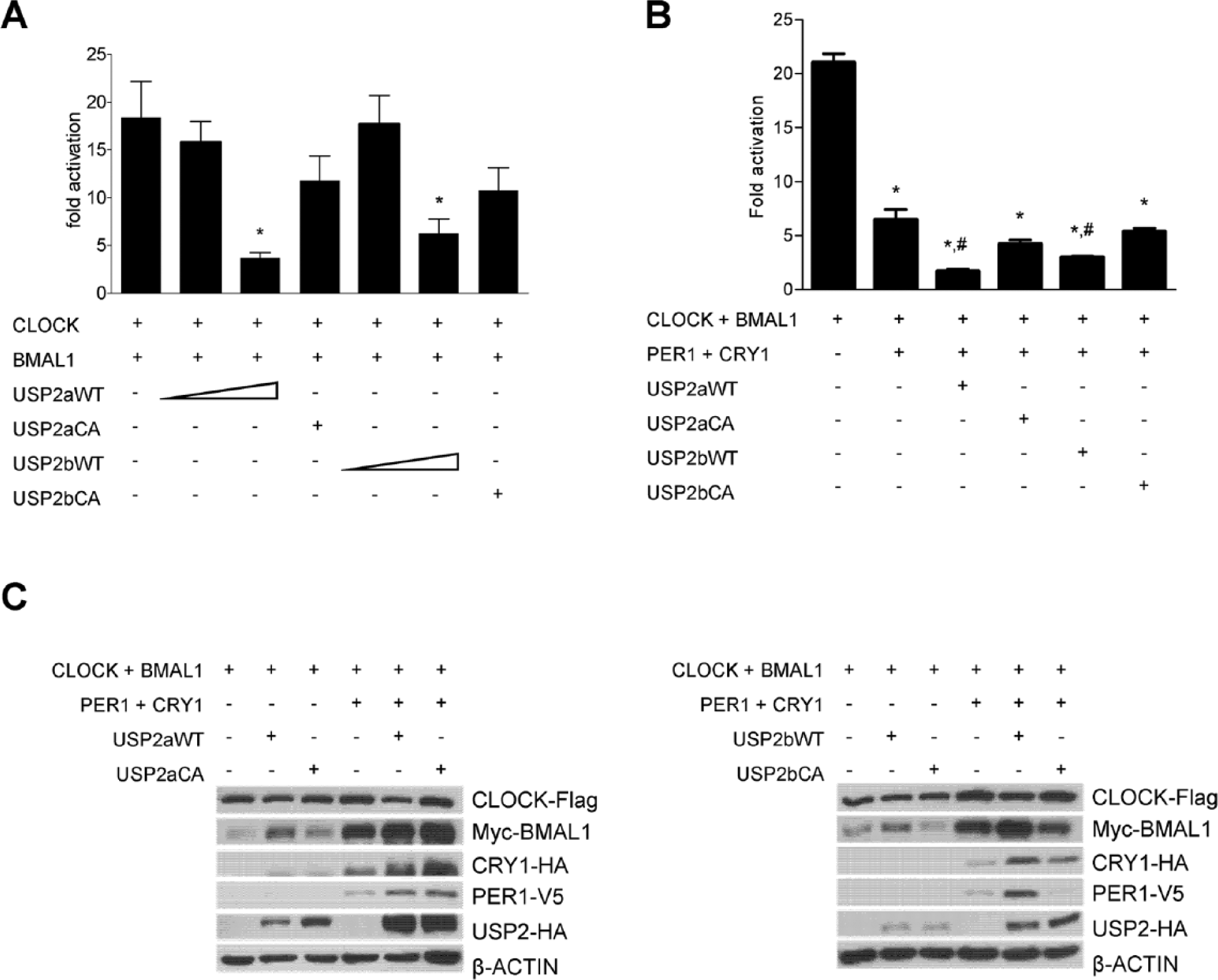
USP2 regulates PER1-mediated repression of CLOCK-BMAL1 transcriptional activity. (A) HEK293 cells were transfected with plasmids expressing CLOCK-Flag, BMAL1-Myc, USP2a-HA, or USP2b-HA (wild-type [WT, 50 or 200 ng plasmid DNA] or catalytically inactive [CA, 200 ng]), as indicated, together with a luciferase reporter under the control of the Per1 promoter. Luciferase activity was measured in the cell lysates and corrected for protein concentration in each sample. Data represent fold induction over the appropriate control condition (empty expression vectors for CLOCK and BMAL). The graph shows the means ± SEM of 4 independent experiments performed in triplicate. *p < 0.05 vs. CLOCK/BMAL1 (1-way analysis of variance [ANOVA] followed by Bonferroni posttests). (B) The experiment was performed as in (A) but with lower levels of USP2 (50 ng plasmid) and plasmids expressing PER1-V5 and HA-CRY1. The graph shows means ± SEM from the combination of 2 independent experiments performed in triplicate. *p < 0.05 vs. CLOCK/BMAL1; #p < 0.05 vs. CLOCK/BMAL1/PER1/CRY1 (1-way ANOVA followed by Bonferroni posttests). (C) Whole-cell lysates were also subjected to immunoblotting with the appropriate antibodies to assess levels of transfected proteins. Shown are blots of a representative experiment from (A), the left panel, and (B), the right panel.
USP2 Regulates the Timing of Nuclear Localization of PER1 in MEFs
A time-dependent, rhythmic nuclear accumulation of PER1 can be observed in NIH3T3 fibroblasts whose clocks have been synchronized by a serum shock (Tamaru et al., 2003). We therefore tested whether USP2 regulates such a rhythm of PER1 nuclear accumulation. In the synchronized WT MEFs, a clear ~24-h rhythm of PER1 nuclear accumulation was observed by confocal microscopy (Fig. 3). Twelve hours (T12) after serum shock, PER1 was detected predominantly in the nucleus. The nuclear PER1 signal then gradually decreased as the cytoplasmic PER1 increased until T20 when PER1 was detected almost evenly in both compartments. Starting at T24, PER1 began to accumulate again in the nucleus, to reach a new peak at T32 (Fig. 3, left panel for PER1 signal, right panel for colocalization with DAPI). In the synchronized Usp2 KO MEFs, a rhythm of PER1 nuclear localization was also observed. However, the phase of PER1 nuclear entry appeared advanced (Fig. 3, compare left and right panels at T28), and the peak nuclear accumulation of the protein was reduced compared with the synchronized WT MEFs (Fig. 3, compare left and right panels at T12 and T32).
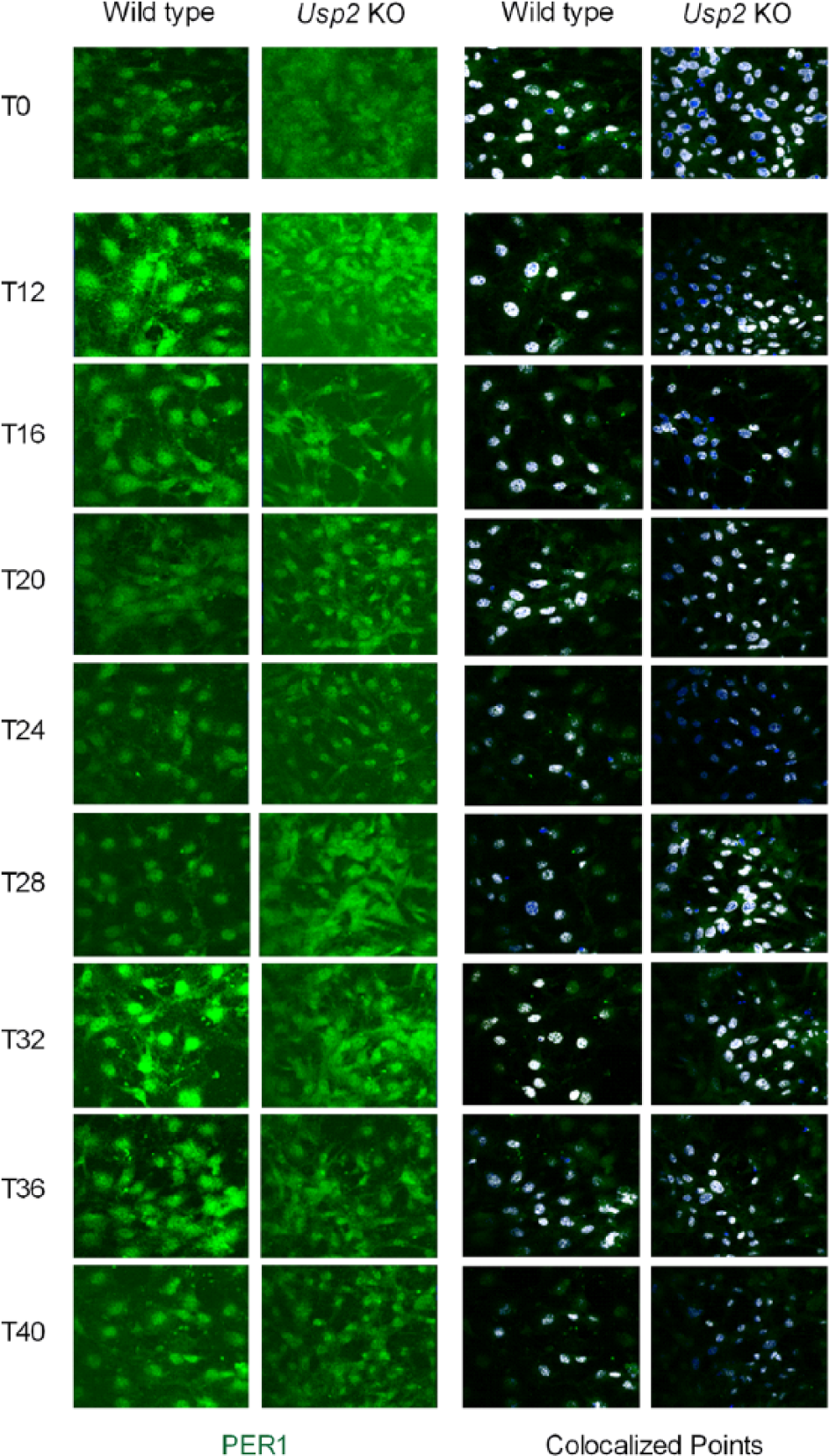
USP2 regulates rhythmic nuclear entry and accumulation of PER1 in synchronized mouse embryonic fibroblasts (MEFs). Wild-type (WT) and Usp2 knockout (KO) MEFs were synchronized by overnight serum starvation followed with 50% serum shock for 2 h. At indicated times (T0 is when horse serum was added), these MEFs were fixed and subjected to immuno-staining for PER1 (green) and DAPI (blue). The nuclear-localized PER1 signals in the series of images over the time course were determined by National Institutes of Health ImageJ colocalization analyses, and colocalized points (white) were identified. Shown is a representative series of images from 3 independent experiments with similar results.
USP2 Is Required for Normal Expression of Clock Genes and Clock-Controlled Genes in MEFs
Since PER1 exerts important regulatory functions in the core clock mechanism, we explored whether the differences in PER1 cycling in Usp2 KO MEFs were associated with differences in clock function. We therefore studied core clock gene RNA expression in synchronized WT and Usp2 KO MEFs that were collected simultaneously with those cells subjected to PER1 immunostaining. Similar patterns of rhythmic expression of Per1, Per2, Bmal1, and Rev-Erbα mRNA were observed in both the WT and KO cells (Fig. 4A). However, the peaks and amplitudes of the core clock gene mRNA levels (except Per1) were significantly decreased or increased in Usp2 KO MEFs (Fig. 4A). We then asked whether this alteration in the core molecular clockwork in Usp2 KO MEFs was associated with disturbed expression of known clock-controlled genes. We examined the mRNA expression of Dbp, Tef, Hlf, and E4bp4 genes, encoding transcription factors that are controlled by the clock. The expression of these clock-controlled genes was dramatically altered in the Usp2 KO MEFs, with a large reduction in Dbp expression and increases in Hlf and E4bp4 expression (Fig. 4B). Tef expression was also significantly altered in KO MEFs but more subtly.
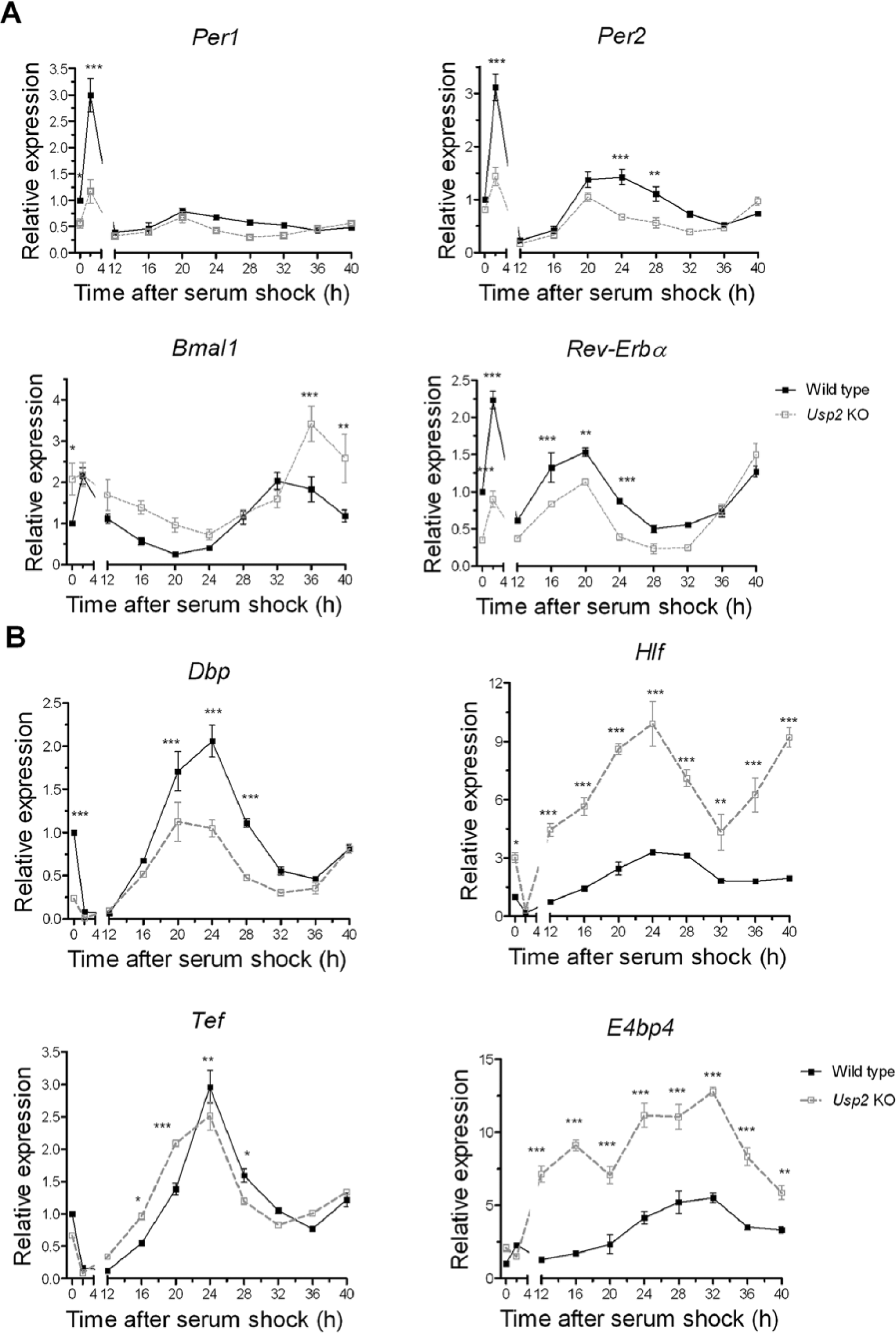
USP2 is required for normal expression of clock genes and clock-controlled genes in mouse embryonic fibroblasts (MEFs). Knockout of USP2 induced significant changes in the rhythmic expressions of core clock genes in MEFs. Wild-type (WT) and Usp2 knockout (KO) MEFs grown and synchronized by serum shock as described in Fig. 3 were collected at the indicated time points. RNA from these cells was analyzed by quantitative polymerase chain reaction for the core clock genes Per1, Per2, Bmal1, and Rev-Erbα (A) and for the clock-controlled genes encoding DBP, TEF, HLF, and E4BP4 (B). Expression was normalized to β-Actin and calibrated to T0 WT MEF samples. Shown are means ± SEM. Two-way analysis of variance for genotype and time, followed by post hoc tests (Bonferroni posttests): effect of genotype, *p < 0.05, **p < 0.01, ***p < 0.001; n = 3 to 5 per time point.
USP2 Regulates the Rhythmic PER1 Nuclear Localization and Clock Gene Expression in the Mouse Liver
We then asked whether USP2 can exert a similar control on PER1 localization and clock gene expression in vivo. Usp2 KO mice and WT littermates were entrained under a 12-h light, 12-h dark cycle and then sacrificed every 3 h over 24 h. Livers were collected for analysis by immunofluorescence for PER1 and quantitative polymerase chain reaction (PCR) for clock gene expression (Fig. 5). A clear nucleocytoplasmic shuttling with a diurnal nuclear entry and accumulation rhythm of PER1 was observed in the WT mouse hepatocytes (Fig. 5A). PER1 was clearly detectable starting at ZT13 in WT mouse hepatocytes in both cytoplasm and nucleus (Fig. 5A, left and right panels). Increased accumulation in the nucleus began at ZT16 and peaked at ZT19. This coincided with the peak levels of USP2 signal in the cytoplasm of hepatocytes (Fig. 5C). Starting at ZT22, the nuclear PER1 signal gradually decreased with a corresponding increase in cytoplasmic PER1 (as observed at ZT1). The total PER1 signal then began to diminish and was undetectable at ZT7. In contrast, in Usp2 KO mouse hepatocytes, the duration of PER1 nuclear localization appeared reduced and its peak phase-advanced. PER1 was detected in Usp2 KO hepatocytes earlier (at ZT10 instead of ZT13) and became undetectable at ZT1 instead of after ZT4 in the Usp2 WT hepatocytes. Although nuclear accumulation of PER1 was observable at ZT19, the PER1 signal appeared weaker in the Usp2 KO hepatocytes. These results indicate that USP2 regulates the diurnal nuclear entry and retention of PER1 in mouse hepatocytes.
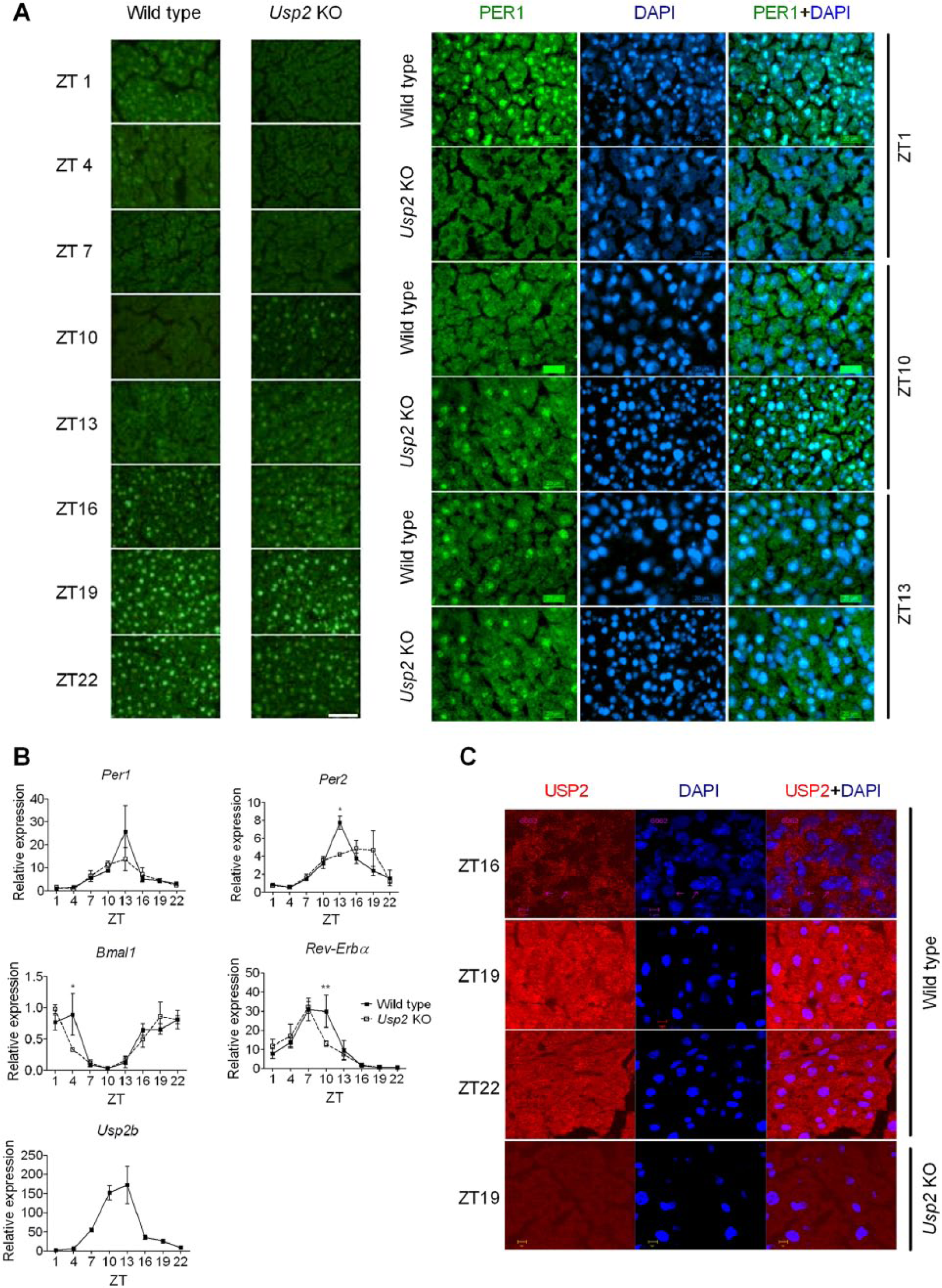
USP2 regulates the rhythmic PER1 nuclear localization and clock gene expression in the mouse liver. (A) Female Usp2 knockout (KO) mice and wild-type (WT) littermates entrained to a 12-h light, 12-h dark cycle were sacrificed every 3 h over 24 h starting at zeitgeber time (ZT) 1. Liver cryosections were immunostained with an anti-PER1 primary antiserum followed by an Alexa Fluor 488 secondary antibody and then counterstained with DAPI (left, 10×; right, 40×). (B) RNA isolated from livers samples described in (A) were analyzed by quantitative polymerase chain reaction for core clock genes Per1, Per2, Bmal1, Rev-Erbα, and Usp2b. Expression was normalized to β-Actin and calibrated to ZT1 WT liver samples. Shown are means ± SEM. Two-way analysis of variance for genotype and time, followed by post hoc tests (Bonferroni posttests): effect of genotype, *p < 0.05, **p < 0.01; n = 3 to 7 per time point. (C) Tissue sections described in (A) were processed for immunostaining of the endogenous USP2 with rabbit anti-USP2 primary antibody (c-terminal L523) followed by an Alexa Fluor 568 goat anti–rabbit antibody and then counterstained with DAPI.
These results prompted us to examine whether loss of USP2 also alters clock gene expression in the liver. Quantitative PCR on liver RNA samples revealed that a rhythmic expression of Per1, Per2, Bmal1, and Rev-erbα mRNAs was observed in both the WT and KO mouse livers (Fig. 5B). As in MEFs, Per1 mRNA levels were not significantly different between WT and KO livers. The peak levels of other core clock gene mRNAs (Per2, Bmal1, and Rev-Erbα) were significantly decreased in Usp2 KO livers (Fig. 5B). The expression profiles of the Per2 and Rev-Erbα were altered in the same directions as in the Usp2 KO MEFs (Fig. 4A and Fig. 5B). These results suggest a similar regulatory role for USP2 in MEFs and in vivo.
Discussion
In this report, we show that the deubiquitinating enzyme USP2 regulates PER1 intracellular localization. In cultured cells, overexpression of USP2 increased the proportion of PER1 found in the nucleus, whereas the loss of USP2 in KO MEFs resulted in higher cytoplasmic localization of the protein. In mouse livers, the timing of PER1 nucleocytoplasmic transfer is altered. This suggests a role for USP2 in regulating the fate of key proteins of the clock feedback mechanism and thus the speed at which the clock runs.
Most roles of ubiquitination in the circadian clock known so far pertain to the regulation of clock protein stability. This is the case for the ubiquitination of PER proteins by β-TRCP–containing cullin ring type E3 ubiquitin ligase complexes, which targets them to proteasomal degradation (Eide et al., 2005; Shirogane et al., 2005; Reischl et al., 2007). This is also true for the destabilization of CRY proteins following ubiquitination by FBXL3-containing SCF complexes (Busino et al., 2007; Siepka et al., 2007). Finally, lithium-induced degradation of REV-ERBα involves the E3 ligases Arf-bp1 and Pam (Yin et al., 2010). So far, the only deubiquitinating enzyme shown to have a role in the clock is USP2. USP2 acts on several clock proteins. First, in cell lines and in liver, USP2 binds and deubiquitinates CRY1, thereby stabilizing it (Tong et al., 2012). Second, USP2 binds to BMAL1 and stabilizes it, and its absence in Usp2 KO mice leads to higher BMAL1 levels (Scoma et al., 2011). In contrast to CRY1 and BMAL1, our previous report showed that USP2 deubiquitinates PER1 without significant effects on the overall stability of this clock protein (Yang et al., 2012). This suggested that deubiquitination of PER1 by USP2 has other consequences than protecting this protein from proteasomal degradation.
PER1 nuclear localization is precisely regulated during the circadian cycle. In each cycle, levels of the protein rise in the cytoplasm, translocate to the nucleus at a precise time, and then return again to low basal levels. Both in MEF cells synchronized by serum shock and in hepatocytes in vivo, we observed that loss of USP2 resulted in a phase advance of this nuclear localization cycle of PER1. Together these findings indicate that USP2 is not essential for nuclear localization or retention but plays a key role in the precise gating of PER1 entry into the nucleus. PER1 shuttling between the cell nucleus and cytoplasm is a dynamic event dependent on its nuclear localization signal and nuclear export signal (Vielhaber et al., 2001). PER1 undergoes phosphorylation in the cytoplasm, and the phosphorylation is required for its translocation. In addition, PER1 was associated with CRY1 and is retained in the nucleus by an undefined mechanism thought to be facilitated by the nuclear export signal of CRY1 (Yagita et al., 2002). However, the same group also found that PER1 can shuttle between the nucleus and the cytoplasm in the absence of endogenous CRY1/CRY2 (Yagita et al., 2000). The precise molecular mechanisms by which USP2 exerts its effect on cellular distribution of PER1 remain unclear. Since USP2 can be localized in the nucleus or in the cytoplasm (Fig 5C and Lin et al., 2000), its effect on PER1 may be occurring in either of these compartments. USP2 did not appear to affect the ability of PER1 to associate with CRY1 when these proteins were expressed in cells (data not shown). However, our results suggest that ubiquitination of PER1 serves as an additional regulatory mechanism for regulating its nuclear entry or retention.
Interestingly, recent studies have indicated a nondegradative role for ubiquitination of CRY proteins. As mentioned above, the cullin ring ligase complex containing FBXL3 ubiquitinates CRY proteins, targeting them to degradation by the proteasome (Busino et al., 2007; Siepka et al., 2007). This is thought to happen in the nucleus and hasten the decline in CRY levels, thus allowing the reinitiation of CLOCK/BMAL1 transcriptional activity (Busino et al., 2007; Siepka et al., 2007; Kurabayashi et al., 2010). Accordingly, in mice with either a loss-of-function mutation or a deletion of the Fbxl3 gene, the free-running period of locomotor activity rhythms and the period of rhythms of SCN tissue in vitro are much longer than in WT controls (Godinho et al., 2007; Siepka et al., 2007; Hirano et al., 2013; Yoo et al., 2013). In sharp contrast, mice lacking FBXL21, a protein closely related to FBXL3, display a short period of locomotor activity rhythms and in vitro SCN rhythms (Hirano et al., 2013; Yoo et al., 2013). Biochemical assays suggest that FBXL21 antagonizes the destabilizing function of FBXL3 and even stabilizes CRY proteins. It may do it by promoting their retention in the cytoplasm but also by binding CRYs and thus preventing them to be ubiquitinated by the FBXL3-containing SCF complex, in both cases delaying the progression of the circadian clock loop (Hirano et al., 2013; Yoo et al., 2013). Therefore, FBXL21 may act in this nondegradative manner in the phase of the circadian feedback loop when CRY1 is accumulating in the cells, while FBXL3’s role would be in the phase of the cycle where CRYs are in the nucleus and then decline in levels. More generally, a model is emerging where PER and CRY proteins are regulated by different posttranslational modifications, acting at different levels of the transcriptional-translational feedback loop. These modifications of the proteins occur in sequential and conditional fashion and can be restricted to specific cell compartments or in time (i.e., the part of the cycle where they either accumulate or decay). On one hand, ubiquitin ligases seem to have very specific roles in the clock, with distinct ligases for specific groups of clock proteins and even distinct ligases at different stages of the feedback loop. On the other hand, deubiquitination in the clock might be less specific, with only one enzyme so far identified to play a role in the clock, USP2. We propose that USP2 may remodel ubiquitin chains on PER1 and thereby affect its nucleocytoplasmic shuttling, a possibility that remains to be explored.
This regulation by USP2 has functional consequences for the clock mechanism. PER proteins interact with CRY proteins to act as transcriptional repressors of CLOCK/BMAL1-driven transcription (Kume et al., 1999). Here we showed that USP2 promotes PER1/CRY1-mediated transcriptional repression. The absence of USP2 (in MEFs or liver) results in altered mRNA expression of many clock genes. Genes encoding clock-controlled transcription factors were also affected. Indeed, in the Usp2 KO MEFs, the mRNA expression of the Dbp, Hlf, and E4bp4 genes was dramatically changed, either increased (Hlf and E4bp4) or decreased (Dbp). DBP and HLF activate and E4BP4 represses Per1 gene transcription through binding to its D-box (Yamaguchi et al., 2000; Doi et al., 2001; Mitsui et al., 2001; Doi et al., 2004; Ueda et al., 2005). The increased expressions of both HLF and E4BP4 with opposing effects on PER1 expression may account for the lack of change in Per1 mRNA levels observed between Usp2 WT and KO cells.
We previously showed that USP2 associates with many components of the core clock complex, but of these proteins, PER1 is the only one it binds directly. Thus, it is conceivable that USP2 mediates the effects on gene expression and intracellular localization through its direct action on the ubiquitinated form of PER1. However, as described above, USP2 can also modulate the ubiquitination of BMAL1 (Lee et al., 2008; Scoma et al., 2011) and CRY1 (Tong et al., 2012). Therefore, we cannot exclude the possibility that the new roles of USP2 in the clock uncovered here are also dependent on its effects on CRY1, BMAL1, or additional substrates yet to be identified. USP2 has multiple substrates in the molecular clock. Moreover, these substrates modulate each other through a web of interacting loops. For these reasons, the implication of USP2 in the clock is complex, and phenotypes of Usp2 KO mice or cells are a sum of the different roles of this enzyme in the clock. Nonetheless, all studies so far, including the new data reported here, point to an important role for the rhythmic expression of USP2 in modulating clock function—in its core mechanism, input pathways, and downstream effects.
Footnotes
Acknowledgements
We thank all members of S.S.W. and N.C.’s laboratories for discussion, as well as K. Stojkovic, G. Baquiran, and N. Bedard for technical help. This work was supported by grants from the Canadian Institutes of Health Research (MOP82734 and MOP115106, to S.S.W.) and the Natural Sciences and Engineering Research Council (249731-2006 RGPIN, to N.C.). N.C. was supported by an FRSQ salary award and a McGill Faculty of Medicine award.
Author Contributions
Y.Y., D.D., N.C., and S.S.W. designed the study and wrote the manuscript. Y.Y. performed all experiments and analyses except those for Fig. 2, which were done by D.D., and the immunostaining and image processing for Fig. 5A, which were done by J.F.
Conflict Of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.