
Abstract
Mitochondria are key players in the generation and regulation of cellular bioenergetics, producing the majority of adenosine triphosphate molecules by the oxidative phosphorylation system (OXPHOS). Linked to numerous signaling pathways and cellular functions, mitochondria, and OXPHOS in particular, are involved in neuronal development, connectivity, plasticity, and differentiation. Impairments in a variety of mitochondrial functions have been described in different general and psychiatric disorders, including schizophrenia (SCZ), a severe, chronic, debilitating illness that heavily affects the lives of patients and their families. This article reviews findings emphasizing the role of OXPHOS in the pathophysiology of SCZ. Evidence accumulated during the past few decades from imaging, transcriptomic, proteomic, and metabolomic studies points at OXPHOS deficit involvement in SCZ. Abnormalities have been reported in high-energy phosphates generated by the OXPHOS, in the activity of its complexes and gene expression, primarily of complex I (CoI). In addition, cellular signaling such as cAMP/protein kinase A (PKA) and Ca+2, neuronal development, connectivity, and plasticity have been linked to OXPHOS function and are reported to be impaired in SCZ. Finally, CoI has been shown as a site of interaction for both dopamine (DA) and antipsychotic drugs, further substantiating its role in the pathology of SCZ. Understanding the role of mitochondria and the OXPHOS in particular may encourage new insights into the pathophysiology and etiology of this debilitating disorder.
Brain development, neuronal plasticity, and synapse connectivity are highly complex processes, related to various brain functions such as learning, memory, emotions, cognition, and sensorimotor function. These core processes facilitate the brain’s constant interaction with and adaption to the environment and are highly dependent on continuous oxygen supply. 1,2 The latter is evident by high energetic demands and oxygen uptake of the brain, roughly 20% of total body consumption while only about 2% of body’s weight. 3 Energy in the form of adenosine triphosphate (ATP) is generated mainly in mitochondria by the oxidative phosphorylation (OXPHOS) process, in which electrons produced by the citric acid cycle are transferred down the mitochondrial respiratory complexes. Mitochondria and the OXPHOS are not only responsible for production of high-energy phosphates, such as ATP and phosphocreatine, but are also involved in a variety of cellular processes, including calcium homeostasis, cAMP/protein kinase A (PKA) signaling, inflammation, reactive oxygen species (ROS) production, and apoptosis. 4 –9 Therefore, it is not surprising that multifaceted OXPHOS dysfunctions, originating from both genetic (maternal, or mendelian inheritance) and environmental influences, have been reported in many diseases and disorders, including neuropsychiatric disorders such as Alzheimer and Parkinson diseases, schizophrenia (SCZ), and bipolar disorder (BD). 2,10 –12 SCZ is a severe mental disorder, heavily affecting the lives of those afflicted and their families. The disorder is characterized by various abnormal cognitive, affective, and motor behavioural features, associated with a variety of impairments in occupational and social functioning. No single symptom is pathognomonic of SCZ; consequently, the disorder is noted for its great heterogeneity across individuals and for its variability within individuals over time. 13 One consistent pathological finding implicated in SCZ is abnormal brain energy metabolism in specific neuronal circuits. Mitochondria, key players in brain bioenergetics, portray various deficits in SCZ. This review focuses on malfunctioning of the OXPHOS, source of ATP production, and main driving force of various mitochondrial-related cellular functions.
Brain Energy Metabolism in SCZ
Brain activity and function depend profoundly on ATP supply, with energetic demands varying significantly according to neuron type and brain activity levels. 6,14,15 Cellular buffering of brain ATP is mainly regulated by ATPase and creatine kinase reactions. 14 Creatine is phosphorylated by creatine kinase and converted into phosphocreatine (PCr), a high-energy phosphate, capable of donating a phosphate group to adenosine diphosphate (ADP), forming ATP, and vice versa. 6 In SCZ patients, most imaging studies using positron emission tomography (PET), functional magnetic resonance imaging (fMRI), magnetic resonance spectroscopy (MRS), phosphorous magnetic resonance spectroscopy ( 31 P-MRS), and single-photon emission tomography (SPECT) reveal altered metabolism, as expressed by changes in glucose, PCr, and ATP in different brain regions, including the prefrontal cortex (PFC), left temporal lobe, and frontal lobe. 16 –22 Interestingly, the severity of negative symptoms and neuropsychological performance correlate with ATP and PCr levels. 23 In BD, which also portrays psychotic symptoms and shares genetic risk with SCZ, 31 P-MRS studies did not find significant alterations in ATP levels. 24,25 However, similar alterations have been reported for both disorders in other brain bioenergetic indicators such as elevated lactate and decreased intracellular pH (pHi) levels, as well as abnormalities in PCr and creatine kinase. 24 –30 It has been suggested that this shift from aerobic respiration to anaerobic glycolysis increases the risk for metabolic syndromes in these patients. 31,32 Such maladaptations to the energetic demands of the central nervous system (CNS) point to a dysfunction of brain mitochondrial OXPHOS. Producing about 90% of ATP molecules generated in the brain, the OXPHOS is responsible for powering cell signaling and neuronal activity processes, such as pre- and postsynaptic action potentials, neurotransmitter release, and postsynaptic currents. 14,33 Thus, damage to the OXPHOS can have detrimental effects on the CNS energetic balance that may lead to various neuronal dysfunctions.
Mitochondrial OXPHOS
The OXPHOS consists of 5 protein complexes and 2 electron carriers embedded in the inner mitochondrial membrane. 34,35 High-energy phosphate production is achieved by coupling electron transfer to proton translocation across the inner membrane, resulting in an electrochemical gradient, which generates a motive force driving ATP synthesis by the fifth complex, ATP synthase (complex V [CoV]). During respiration, electrons are first transferred from the citric acid cycle products, NADH and succinate, through complexes I (CoI) and II (CoII), respectively, to ubiquinone. They then pass through complex III (CoIII) and cytochrome c, terminating at complex IV (CoIV). In this process, CoIV reduces O2 to H2O. 35 The interaction between OXPHOS complexes and their organization is still unclear. According to the “fluid-state model,” these complexes diffuse freely across the membrane, transferring electrons by randomly colliding. In contrast, the “solid-state model” supports the formation of stable supercomplex assemblies, or respirasomes, composed of several complexes functioning together. 36,37 The assembly process of the OXPHOS complexes is tremendously intricate and is composed of multiple stages in which many structural, catalytic, and assembly proteins participate. In addition, proteins encoded by nuclear DNA (nDNA, ∼70 proteins), synthesized in the cytosol and imported into the mitochondria, have to assemble with proteins encoded by mitochondrial DNA (mtDNA, 13 proteins). 38 Deficiencies in OXPHOS complexes formation and function are associated with either mtDNA or nDNA mutations and can lead to various defects, including synapse damage, axon degradation, ROS production, apoptosis, and cell death. 4,39 –41
Functional and Genetic Alterations of the OXPHOS in SCZ
Many patients diagnosed with mitochondrial diseases with a variety of OXPHOS deficiencies, CoI in particular, portray psychotic-like symptoms. 12 On the other hand, OXPHOS dysfunction has been widely reported in disorders with psychotic features, such as SCZ and BD. 42 –45 Taken together, these findings suggest a role for mitochondrial dysfunction in the pathophysiology of psychosis. Dysfunctions of the OXPHOS have been observed at the level of high-energy phosphate production as well as enzymatic activity and subunit expression of OXPHOS complexes. Genetic studies implicate OXPHOS complexes, particularly CoI, in both disorders. 23,27,44,46 –50
Functional impairments of OXPHOS complexes I, II, and IV have long been described in various brain areas of SCZ patients. 47 –49,51 In BD, however, only 1 study has reported a decrease in CoI activity in the PFC, with no change in SCZ patients. 52 We have reported aberrant OXPHOS activity in platelets of SCZ patients, with CoI showing the most consistent impairments. Our studies show that in blood cells, the enzymatic activity of CoI, but not that of complexes I to III and CoIV, exhibits disease state–dependent alterations. Thus, increased CoI activity positively correlates with positive symptomology and active psychosis, while decreased activity is associated with the residual state. 51 Transcriptomic, proteomic, and metabolomic studies in different brain areas, mostly the PFC, have demonstrated specific robust changes (both increases and decreases) in gene expression and protein level associated with mitochondrial function in SCZ, among them OXPHOS genes and proteins. For example, the expression levels of several CoI subunits, such as NDUFV1, NDUFV2, and NDUFS1, which comprise one functional subunit that includes CoI’s substrate binding site, were significantly altered in the PFC, striatum, hippocampus, and parieto-occipital cortex as well as in somatic cells. 27,53 –58 Interestingly, 1 study reported a positive correlation between the extent of change in blood cells and positive symptomology in SCZ first-episode patients. 50 In BD, transcriptomic abnormalities of mitochondrial genes were also reported mainly in subunits of complexes I, IV, and V. 59,60 However, both disorders differ with respect to the extent and characteristics of mitochondrial impairments, energy metabolism, and ROS production. Overall, there is an inconsistency in the literature regarding alterations in OXPHOS gene expression. These discrepancies may be explained in part by our findings that the change in gene expression exhibits a disease-specific neuroanatomical pattern 44 and/or is cell type specific. 56 Moreover, varying results may be due to disease subtype and ethnicity of the tested population, as has been shown for NDUFV2 protein expression in lymphoblastoid cells of Japanese and European Caucasian BD-I and BD-II patients. 58,61 We have previously suggested that change in expression of at least some of the mitochondrial genes in SCZ can be attributed to abnormal expression of the transcription factor Sp1, which controls many nDNA encoded mitochondrial genes, including NDUFV2 and NDUFV1. 62 Indeed, a highly significant positive correlation and a parallel pattern of change in transcripts of Sp1 and CoI subunits in various brain areas and blood cells have been observed. 62
A growing number of studies have found susceptibility loci and associated risk genes, including those of the OXPHOS in SCZ, even though genome-wide association studies (GWAS) have failed to consistently replicate genetic risk factors among these genes. 63 –65 One of these NDUFV2 has been reported to be a high-risk gene for SCZ. 66 Single-nucleotide polymorphisms (SNPs) in its promoter, introns, and 3′-UTR regions, have been associated with both SCZ and BD. 67,68
A small but growing number of studies report mtDNA SNPs as risk factors in SCZ, 69,70 possibly due to somatic rather than inherited mutations. 29 SNPs in ND1, ND4, and ND5 mtDNA encoded subunits of CoI have been repeatedly reported in SCZ. 71,72 In line with the latter, a significant comorbidity was observed between SCZ and mitochondrial disease. 12,73 Taken together, the data obtained from functional, expression, and genetic evidence point to a role for OXPHOS complexes, particularly CoI, in SCZ. In this context, it is noteworthy that CoI has been shown to be the rate-limiting enzyme for O2 consumption in nerve terminals, as a 10% inhibition of CoI is sufficient for major alterations in O2 consumption. 74
The OXPHOS and cAMP/PKA Signaling
Driving cellular respiration, on one hand, and mitochondrial apoptosis cascade, on the other, the OXPHOS is linked to different, tightly regulated cell signaling pathways, with cyclic AMP (cAMP) and its effector PKA being the most studied OXPHOS-related cellular signaling pathway. 8,75 The significance of this pathway in cell growth, survival, neuro-protection, axon regeneration, and ROS production is well established. 76 –78 However, relatively little is known about the mechanisms of action of mitochondrial cAMP and PKA. 8,75 In mitochondria, it has been shown that this pathway plays a role in fission/fusion process via phosphorylation and inactivation of the fission protein dynamin-related protein 1 (Drp1), thereby promoting mitochondrial elongation and facilitating fusion, which is important for neuronal survival. 79,80 Identification of cAMP and soluble adenylyl cyclase (sAC) inside the mitochondria indicates the local production of mitochondrial cAMP. 8,81 PKA, the downstream target of cAMP, has also been shown to be located inside the mitochondria at different compartments (outer membrane, intermembrane space, and the matrix), despite not having a mitochondrial targeting sequence. 8 In line with these findings, cAMP/PKA signaling has been shown to regulate transcription and phosphorylation of OXPHOS subunits from both nuclear and mitochondrial origin. 8,75,82 cAMP/PKA-dependent phosphorylation of nuclear CoI encoded subunits has been reported for NDUFB11, NDUFA1, NDUFS4 NDUFA7, NDUFA10, NDUFC2, and GRIM19. 83 –87 Interestingly, 1 study has shown that phosphorylation of NDUFB11 results in a reduction of CoI activity. 88 In line with the latter, inhibition of sAC, which product cAMP was shown to prevent degradation of CoI subunits NDUFS4, NDUFS2, and NDUFA9, reduced CoI activity. 89 A tissue-specific activity of the cAMP/PKA pathway was also shown to regulate CoIV activity by phosphorylation of various subunits. 90,91 This interaction is further exhibited by the involvement of targets downstream to cAMP/PKA, such as cAMP-responsive element-binding protein (CREB) and peroxisome proliferator–activated receptor coactivator 1α (PGC-1α), in the transcription of mitochondrial genes from both mitochondrial and nuclear origin. 82,92,93 CREB, in addition to its functions in the nucleus, is imported into the mitochondria via the translocase of the outer membrane (TOM complex). In the mitochondria, CREB was shown to regulate the expression of mtDNA encoded genes, ND1, ND6, and COXIII in the periphery and ND2, ND4 and ND5 in brain. 94 In brain, CREB induced changes in the mtDNA encoded genes affecting CoI activity and CoI-dependent respiration. 94 PGC-1α activates transcription factors regulating the transcription of OXPHOS subunits by Sp1 and nuclear respiratory factors (NRF1 and NRF2). 95,96 cAMP/PKA signaling pathway also affects cellular calcium homeostasis, which is discussed in the following section.
Abnormalities in the cAMP/PKA signaling pathway have been reported in SCZ. For example, postmortem analysis of SCZ patients revealed an asymmetry of cAMP binding to PKA in temporal cortices. 97 An alteration in PKA activity has been reported in the dorsolateral prefrontal cortex (DLPFC) but not in the anterior cingulate (ACC) in SCZ. No changes were observed in levels of PKA catalytic subunits. 98 A more significant change in the cAMP/PKA signaling pathway was observed in BD, with a widespread decrease in cAMP binding to PKA in brain 99 and abnormal PKA level and activity. 100,101 In platelets of both SCZ and BD patients, abnormal levels of the catalytic subunits of PKA were reported. 102,103 Alterations in CREB expression and protein levels were also reported to vary according to brain region, in a disease-specific manner, with a decrease in the cingulate gyrus (CG) and in the DLPFC in BD and a decrease in the CG with no change in the DLPFC in SCZ. 104 Finally, a number of phosphodiesterases (PDEs) have been shown to inactivate cAMP by hydrolysis. 105 –108 PDE4 has been shown to interact with the protein disrupted in schizophrenia 1 (DISC1) in a cAMP/PKA-dependent manner and inactivate cAMP. 109 Both proteins have been localized to the mitochondria, 110 suggesting that variations in both can affect mitochondrial cAMP catabolism, with elevated cellular cAMP in SCZ leading to dissociation of PDE4B from DISC1 and increased PDE4B activity. 109 All 3 proteins have been implicated in SCZ. For example, alterations in PDE4A and PDE10 protein levels have been observed in different brain regions of SCZ patients, and SNPs were identified in several PDEs and in DISC1. 66,111 –113 These findings attribute an additional role for cAMP/PKA signaling pathways in the interaction between mitochondria and the cell, regulating mitochondrial activity according to cell demands and vice versa.
The OXPHOS and Calcium Homeostasis
Calcium, one of the cells’ most common second messengers, is involved in many essential cellular functions, including gene transcription, signaling pathways, cell proliferation and regulation of neuronal functions, synaptic plasticity, learning, memory, and cognition. Mitochondria play an important role in Ca+2 buffering and signaling, while Ca+2 regulates mitochondrial localization, movement within the neuron, and their degradation.
114
–117
N-methyl-d-aspartate (NMDA) receptors (NMDARs) are responsible for the main flux of Ca+2 into CNS cells.
30
Cellular Ca+2 is mainly stored within the endoplasmic reticulum (ER), where its concentration is several orders of magnitude higher than in the cytoplasm.
118,119
ER and the mitochondria connect and interact via the mitochondria-associated ER membrane, allowing release of Ca+2 at maximal proximity.
120,121
The main mechanism of ER calcium release is through inositol-1,4,5-trisphosphate (IP3), which activates the IP3 receptor (IP3 R) on the ER membrane, leading to Ca+2 release into the cytosol.
122,123
The cAMP/PKA pathway plays an important role in Ca+ signaling, with PKA modulating both IP3 R capacity
124
and NMDAR permeability for extracellular Ca+2.
30
Cytosolic Ca+2 enters the mitochondria via the Ca+2 uniporter, due to the membrane potential driving force generated by the OXPHOS.
123,125
Intramitochondrial Ca+2 (
The OXPHOS in Neuronal Development and Plasticity
Adaptation of the nervous system to the ever changing environment by neurogenesis and active modulation of synaptic connections between neurons is a high-energy demanding process, termed synaptic plasticity, a concept initially proposed by Donald Hebb in 1949. 151 Defects in neuronal connectivity, synaptic modeling, and neuronal signaling have been suggested to be part of the underling pathophysiological mechanisms of SCZ. 43,152 –156 Mitochondria, localized in dendrites and axons, participate in essential processes related to plasticity, including morphological changes such as development of new synapses and remodeling of mature ones, Ca+2 signaling, generation of action potential, synaptic transmission, and ion homeostasis. 157 –159 Attached to vesicles, they are transported along microtubules to synaptic terminals, enabling these high-energy demanding processes. 160,161 Removal of mitochondria from nerve endings can lead to abnormal synaptic transmission. 162,163 Interestingly, DISC1, implicated in SCZ, affects mitochondria localization and microtubule transport. 164
In cells, mitochondria routinely fuse, divide (fission), branch, and change their size in a dynamic manner. This process, termed mitochondrial network dynamics, enables proper mitochondrial function, including inheritance and maintenance of mtDNA, regulation of metabolic energy, mitochondrial trafficking, and maintenance of a healthy mitochondrial population. 159,165 –167 Mutations related to this process have been previously linked to neurodegenerative diseases such as Parkinson and Huntington diseases, and more recently, impairments in mitochondrial network dynamics have been reported by us in SCZ and by others in BD-derived cells. 168 –173 The extent to which the OXPHOS affects neuronal branching and plasticity is still an open question. However, in CoI mutagenized Caenorhabditis elegans, an increased number of dendrites and their branching in sensory neurons were observed. 174 In humans, we have demonstrated impairments in differentiation and maturation into dopaminergic and glutamatergic neurons of SCZ-derived induced pluripotent stem cells (iPSCs), alongside a reduction in CoI-driven respiration. In addition, dissipation in mitochondrial membrane potential, impaired mitochondrial network structure and connectivity, and abnormal expression levels of NDUFV1, NDUFV2, and NDUFS1 were reported. 170 Mammalian embryonic stem cell (ESCs), which originate from the blastocyst inner cell mass, are naturally exposed to hypoxic conditions, 175 with mitochondria showing immature morphology at this stage. 176 –178 Not surprising, the main source of energy at this stage comes mainly from glycolysis and not oxidative phosphorylation. Only later during differentiation, O2 levels rise and an increase in mitochondria number is accompanied by a shift towards oxidative phosphorylation respiration. 177,179 Indeed, it was shown that cellular differentiation of ESCs and iPSCs depends on OXPHOS and is hampered by the inhibition of CoI or CoIII. 180 –182 In line with these findings, we have shown that abnormalities in mitochondrial function are associated with a failure of SCZ-derived iPSCs to differentiate into dopaminergic and glutamatergic neurons. 170 These findings imply a key role for mitochondria and their OXPHOS in synaptic plasticity and differentiation into neurons.
Dopamine and Antipsychotic Drugs Interact with the OXPHOS
Dopamine (DA) has been suggested to play a pivotal role in the pathophysiology of a number of mental disorders, particularly in SCZ. 183 –186 The DA hypothesis in SCZ originally stemmed from the ability of antipsychotic drugs to inhibit DA receptors, primarily D2 receptors, and that of psychostimulants to activate DA transmission. 187 Even though the mechanisms of action of antipsychotic drugs are not entirely clear, a reduction in energy-demanding processes induced by these substances in the frontal lobes and basal ganglia of medicated SCZ patients has been reported. 23,188 Concomitantly, in vitro exposure of rat pancreas cells to clozapine resulted in reduced levels of glucose oxidation and ATP production. 189 Similarly, inhibition of ATP-related responses was demonstrated following exposure of PC12 cells to haloperidol. 190 We and others have shown that both typical and atypical antipsychotic drugs inhibit CoI activity and CoI-driven respiration in isolated mitochondria and in intact neuronal cells. 191 –194 In mice, acute and chronic haloperidol administration specifically inhibited CoI in extrapyramidal brain regions, the extent of inhibition correlating with D2 abundancy. 195 In human and rat brain specimens, a drug- and dose-dependent in vitro inhibition of CoI activity was observed with haloperidol > chlorpromazine > risperidone > zotepine > clozapine. 169,194,196 Interestingly, we have shown that CoI activity is increased in peripheral blood cells of medicated and nonmedicated SCZ patients at the acute stage, while decreased in chronically medicated SCZ patients at the residual state. 169 Similar to antipsychotics, DA also affects mitochondrial functions. 197 In neuronal cell cultures, L-3,4-dihydroxyphenylalanine (L-DOPA) and DA reduced striatum CoI activity and ATP production. 198 –201 We have shown that these effects on mitochondria are due to the ability of DA to be taken up by the mitochondria and elicit a dose-dependent inhibition of CoI activity but not that of complexes II, IV, and V. 201 Although DA and antipsychotics both inhibit CoI activity, they interact with the complex at different sites, DA with the hydrophilic matrix penetrating arm and antipsychotics with the hydrophobic inner membrane embedded arm of the complex. 169 The clinical efficiency of antipsychotic drugs has been attributed to their antagonism of the D2 receptor, while in the mitochondria, these drugs mimic DA action on CoI. This drug-mitochondria interaction may be one mechanism involved in the side effects exhibited by the drugs. Alternatively, DA and antipsychotic drugs may both interact independently with the mitochondria, participating in a compensatory mechanism aimed to overcome mitochondrial dysfunction.
Conclusion
Neuronal energetic demands render them heavily dependent on the mitochondria, particularly on the OXPHOS. Apart from ATP production, the OXPHOS is a major player in many cellular processes, including calcium buffering, cell signaling, ROS production, and apoptosis. In mental disorders, mitochondrial deficits are significant yet of a relatively limited magnitude. Therefore, a deviation from normal functioning rather than lack of functioning and cell death is expected, specifically in cells highly dependent on energy supply for their activity. Indeed, in mental disorders, mild alterations in brain development, synaptic plasticity, and neuronal network connectivity have been observed. Such changes, however, can affect brain functioning, which may ultimately manifest in distorted cognitive and emotional behaviours, characteristic of mental disorders. As expected, many studies have found defects in various components of the OXPHOS protein apparatus (Figure 1). The fact that the OXPHOS interacts with antipsychotic drugs (typical and atypical) and with dopamine suggests mitochondria in general and CoI in particular, as an additional pathological factor in SCZ, which may serve as a novel target for future treatment strategies.
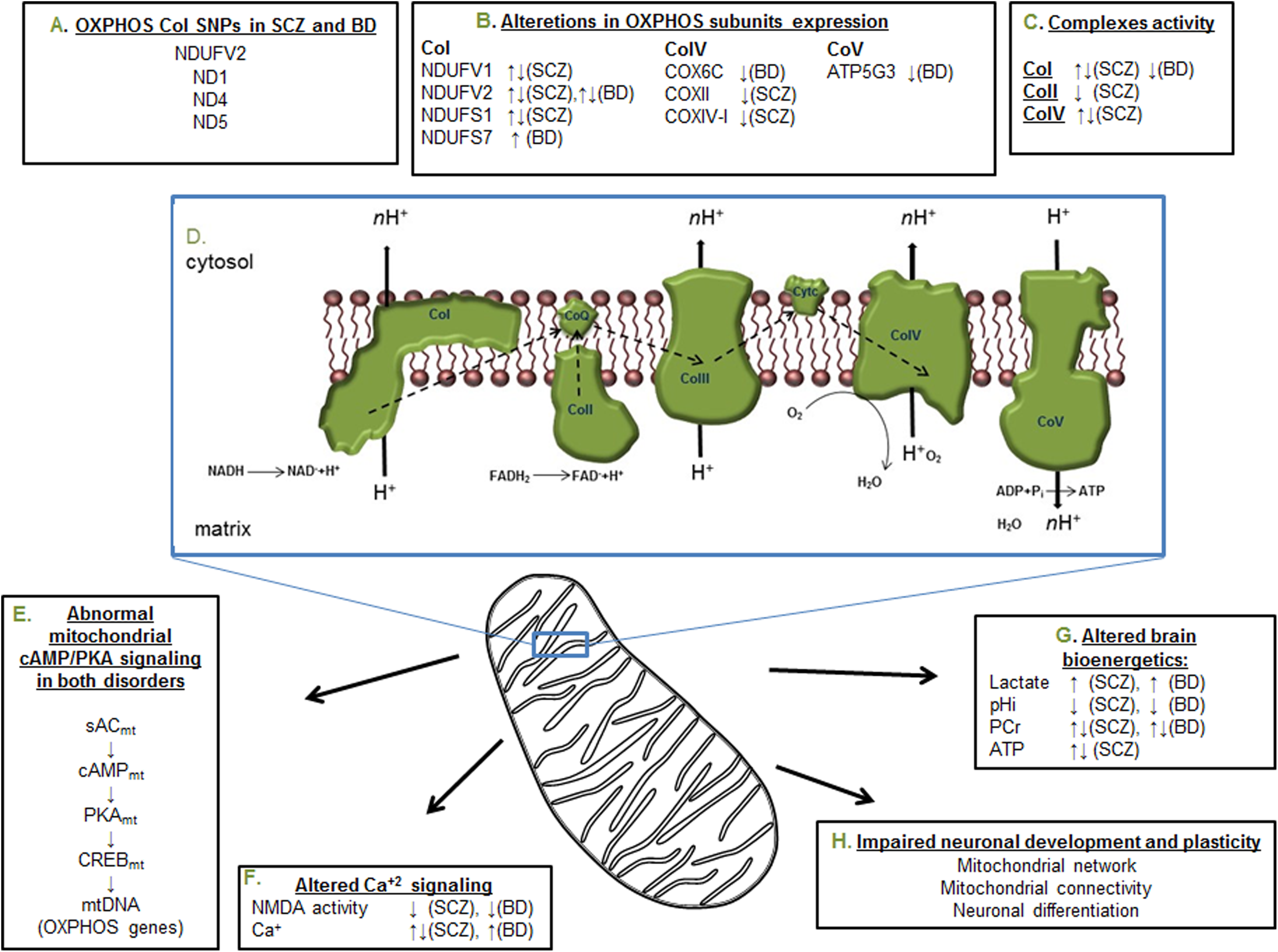
Summary of the most reproducible deficiencies in the oxidative phosphorylation system (OXPHOS) and its related cellular signaling in schizophrenia (SCZ) and bipolar disorder (BD). (A) The most frequent single-nucleotide polymorphisms (SNPs) reported in nuclear and mitochondrial DNA (nDNA and mtDNA, respectively) encoded subunits of complex I. (B) Increase and decrease in the expression of various subunits of the OXPHOS complexes. (C) Reduced and enhanced enzymatic activity of 3 complexes of the OXPHOS. (D) The respiratory chain complexes, electron transfer, and adenosine triphosphate (ATP) production. (E) The mitochondrial cAMP/protein kinase A (PKA) signaling pathway, which affects the expression of mtDNA encoded subunits of the OXPHOS complexes. (F) Altered glutamate NMDA receptor transmission and intracellular Ca2+ concentration and signaling. (G) Alterations in mitochondrial originated high-energy phosphates, lactate, and pH, indicating impaired energy production in cell or tissue. (H) Disease-related neurodevelopmental consequences of the alterations presented in A to G. Arrows indicate the direction of alteration. PCr, phosphocreatine.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.