
Abstract
Mitochondrial dysfunction is commonly observed in bipolar disorder (BD) and schizophrenia (SCZ) and may be a central feature of psychosis. These illnesses are complex and heterogeneous, which is reflected by the complexity of the processes regulating mitochondrial function. Mitochondria are typically associated with energy production; however, dysfunction of mitochondria affects not only energy production but also vital cellular processes, including the formation of reactive oxygen species, cell cycle and survival, intracellular Ca2+ homeostasis, and neurotransmission. In this review, we characterize the upstream components controlling mitochondrial function, including 1) mutations in nuclear and mitochondrial DNA, 2) mitochondrial dynamics, and 3) intracellular Ca2+ homeostasis. Characterizing and understanding the upstream factors that regulate mitochondrial function is essential to understand progression of these illnesses and develop biomarkers and therapeutics.
Highlights
Mitochondrial dysfunction is frequently reported in both bipolar disorder (BD) and schizophrenia (SCZ).
This may lead to increased generation of reactive oxygen species (ROS).
ROS may react with cellular macromolecules to alter signaling pathways, decompensate myelin, and cause damage to DNA and RNA.
Mutations in mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) may produce mitochondrial dysfunction in BD and SCZ.
Disrupted mitochondrial fusion, fission, and trafficking may impair mitochondrial function in SCZ.
Enhanced release of Ca2+ from the endoplasmic reticulum in BD and SCZ may lead to mitochondrial Ca2+ overload, disrupting function.
Introduction
Bipolar disorder (BD) and schizophrenia (SCZ) are severe psychiatric disorders with a lifetime prevalence exceeding 3% of the population worldwide. 1 These disorders are characterized by clinical features such as mania and depression in BD or hallucinations and delusions in SCZ. Relapse and recurrent psychosis are common to both disorders, causing lifelong disease burden and impairment. 2,3 Understanding the etiology and pathophysiology of these disorders is necessary to develop biomarkers and rational therapeutics to ease their burden. Substantial evidence exists suggesting a role for mitochondrial dysfunction in the pathophysiology of major psychoses. 4 –6 Although mitochondria are traditionally associated with adenosine triphosphate (ATP) production, they are also crucial in regulating cell cycle, 7 death and survival, 8 intracellular Ca2+ homeostasis, 9 and neurotransmission. 10 The brain in particular is affected by dysfunction in mitochondria due to its high energy demands and sensitivity to oxidative damage. 11 Because neurotransmitter release and cell survival are dependent on ATP production and Ca2+ homeostasis, mitochondrial dysfunction can alter synaptic connectivity, which may in turn produce symptoms of psychosis. 12,13 Understanding mitochondrial function will be crucial to comprehend disease progression and to develop rational therapeutics to improve the quality of life of patients with psychiatric illness.
Three of the major upstream pathways that may impair mitochondrial function in BD and SCZ include 1) mutations in nuclear and mitochondria DNA, 2) altered mitochondrial dynamics, and 3) perturbed Ca2+ flux. BD and SCZ are complex diseases that cannot be characterized by a singular narrow pathway. Rather, numerous subtle alterations likely converge upon particular pathways (i.e., mitochondrial function) to produce functional alterations. Thus, examining upstream pathways that control mitochondrial function will lead to a more comprehensive understanding of the etiology and pathophysiology of BD and SCZ. In this review, will address how these interrelated upstream processes may contribute to mitochondrial dysfunction in major psychosis.
Mitochondrial Dysfunction in Major Psychosis
Mitochondria are a major endogenous source of reactive oxygen species (ROS). 14 During normal mitochondrial metabolism, only a small proportion of electrons escape the electron transport chain (ETC), mainly through complex I. 15 These electrons reduce O2 to produce superoxide anion, which is then dismutated by superoxide dismutase (SOD) to yield hydrogen peroxide (H2O2) (Figure 1B). 16 In the presence of reduced transition metals, H2O2 may further react to form a hydroxyl radical. 16 Hydroxyl radicals are highly reactive and can oxidize nucleic acids, lipids, and proteins (Figure 1C). 17,18 ROS are strong oxidants and important signaling molecules, whose effects are balanced by antioxidants, 19,20 such as glutathione, SOD, and glutathione peroxidase (GPx). 21 Dysregulation of the ETC may lead to greater proportion of electrons escaping and forming ROS. 22 When ROS production exceeds the capacity of antioxidant networks, the cell is subjected to oxidative stress. 15,18 The brain in particular is susceptible to oxidative stress due to its high-energy demand, easily oxidized polyunsaturated fatty acids, and relatively low antioxidant capacity. 23,24 Compromised mitochondrial function can disrupt neuronal oxidative metabolism. This may alter neurotransmission and neuronal growth, 2 highly energy-dependent processes, producing symptoms of psychosis and altered mood. 15,25
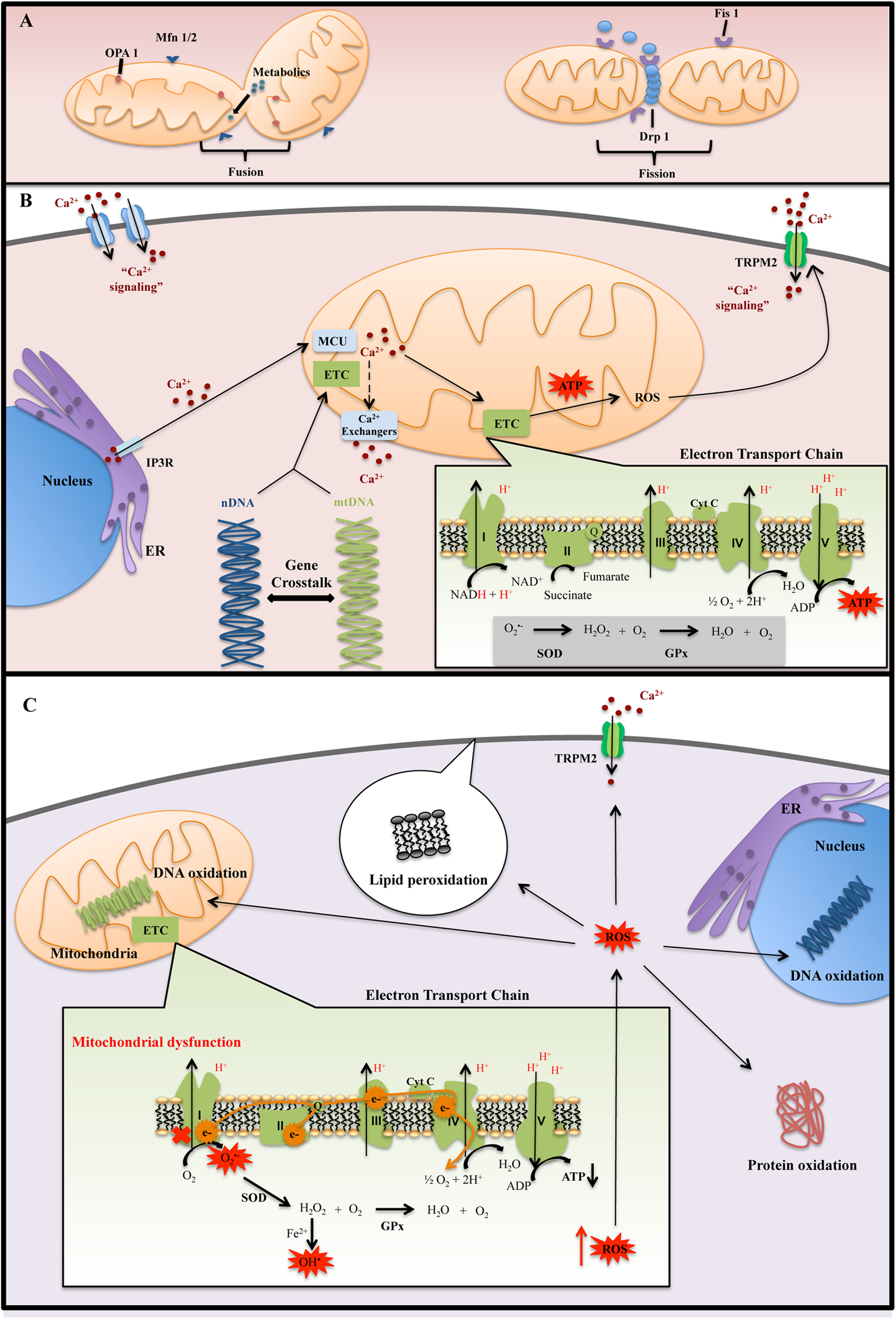
(A) Mitochondrial dynamic: fusion process is important for mitochondrial function by diffusion of metabolites and enzymes between mitochondria, as well as dilution of damaged proteins and DNA. The fusion mediators are Mfn1 and Mfn2, which is present on the outer mitochondrial membrane, and Opa1, which is located in the inner mitochondria membrane. Fission process can isolate injured mitochondria, contributing to mitochondrial quality control. The fission mediators are Fis1 and Drp1. Fis1 recruits Drp1 to mitochondria, and it permits the development of fission process. (B) Normal mitochondrial function: mitochondrial and electron transport chain (ETC) assembly and function are dependent on nuclear DNA (nDNA) and mitochondrial DNA DNA (mtDNA)–encoded proteins. nDNA-encoded proteins regulate mitochondrial replication, transcription, and repair, allowing for crosstalk between nDNA and mtDNA. Mitochondria take up Ca2+ primarily through mitochondrial Ca2+ uniporter (MCU). Ca2+ is then extruded from mitochondria through ion exchangers that are coupled to adenosine triphosphate (ATP) production. Mitochondria are localized close to sites of Ca2+ entry, such as the endoplasmic reticulum (ER) and membrane channels, allowing them to buffer cytosolic Ca2+ concentrations. (C) Mitochondrial dysfunction: ETC impairment increases the amount of electrons leakage, resulting in increased reactive oxygen species (ROS) production. High levels of ROS coupled with low antioxidant defenses disrupt redox homeostasis, leading to cellular oxidative stress. Antioxidant defenses include superoxide dismutase (SOD) and glutathione peroxidase (GPx). If these high levels of ROS are not sufficiently detoxified by these antioxidant enzymes, it can cause oxidative damage to proteins, lipids, and nucleic acids.
Complex I is the first structure of the ETC that catalyzes the transfer of electrons from NADH to ubiquinone 26 and is also the major site of ROS generation in the ETC. 27,28 Dysfunction of mitochondrial complex I is a commonly observed phenomenon in BD and SCZ. A recent review of microarray studies found a consistent downregulation of genes encoding subunits of complex I, including NDUFV1, NDUFS1, NDUFS8, and NDUFS7, in postmortem frontal cortex (PFC) and hippocampus samples of subjects with BD. 4 The subunits that were found to be downregulated in BD formed the catalytic core of complex I involved specifically in electron transfer from NADH to ubiquinone, 29 suggesting that patients with BD may be susceptible to electron leakage through complex I. In contrast, patients with SCZ presented with inconsistent alterations, which included increased and decreased gene expression levels of both structural and catalytic subunits. 4 In agreement with these findings, a previous study demonstrated decreased NDUFS7 protein levels in the PFC of subjects with BD but not SCZ, which were associated with decreased complex I activity. 30 Interestingly, the mood stabilizer Li+ was found to increase expression of complex I subunits in postmortem brains and activity level in vivo of subjects with BD. 31,32 These findings suggests that while complex I dysfunction is present in both disorders, impairments in electron transfer may be more specific to BD (Table 1).
Summary of Pertinent Findings Discussed of Mitochondrial-Related Alterations in Patients with BD and SCZ.
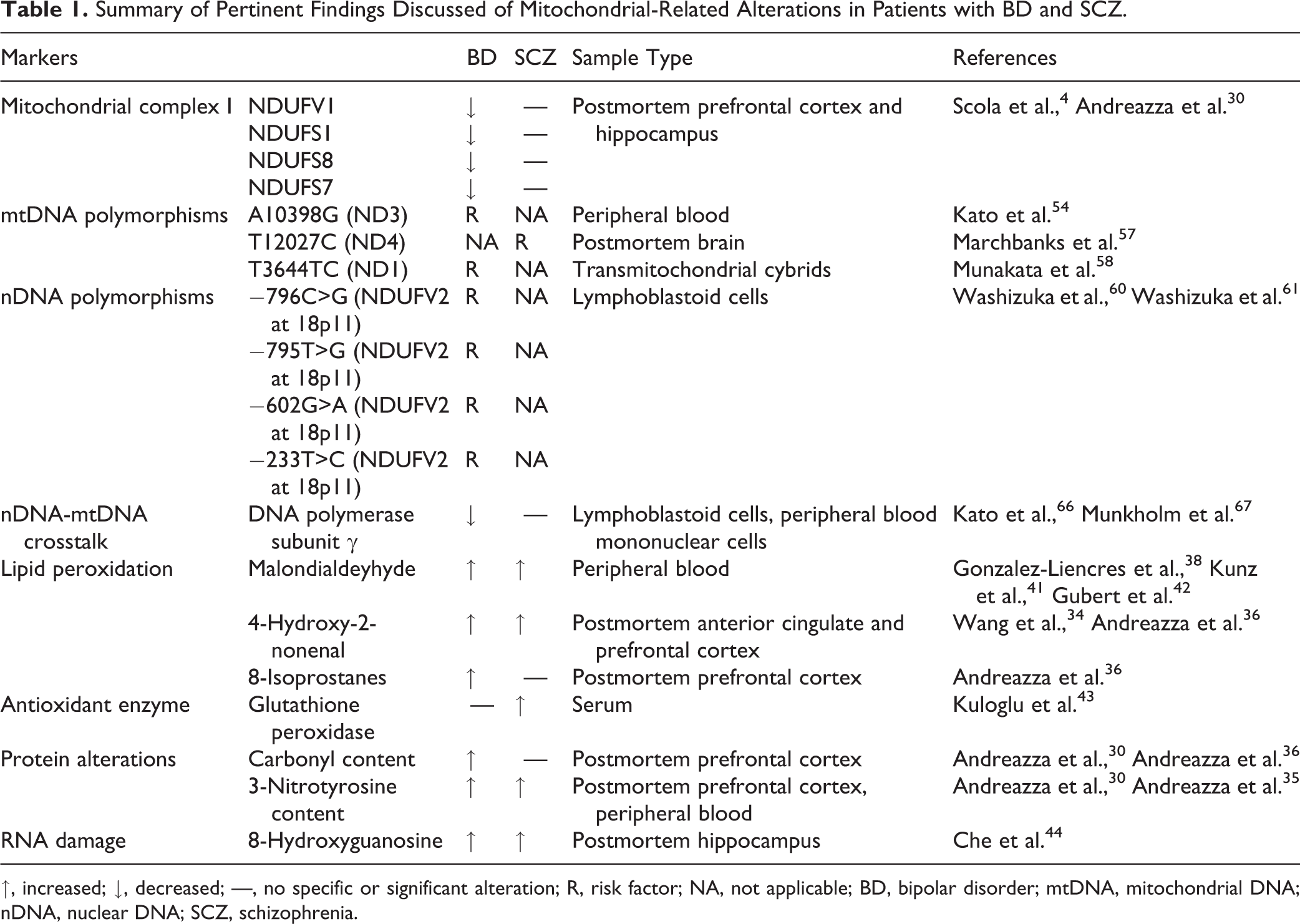
↑, increased; ↓, decreased; —, no specific or significant alteration; R, risk factor; NA, not applicable; BD, bipolar disorder; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; SCZ, schizophrenia.
A major consequence of complex I dysfunction is the generation of ROS, leading to oxidative damage to macromolecules. A number of studies have identified increased oxidative damage markers to protein, lipid, and DNA in BD and SCZ. 30,33 –38 ROS-induced oxidative damage to lipids, for instance, results in lipid hydroperoxides (LPH), which are unstable and react with other lipids to form products such as 8-isoprostanes (8-ISO), malondialdehyde (MDA), 4-hydroxy-2-nonenal (4-HNE), and acrolein. 39,40 MDA has been frequently found to be elevated in both BD and SCZ 41 –43 and was negatively correlated with complex I activity. 42 Oxidative damage to fatty acids, which make up myelin, may cause degeneration of white matter tracts and produce abnormalities in neural circuits. 40 Indeed, elevated levels of 4-HNE have been found in myelin fractions from patients with BD and SCZ, with elevated levels of 8-ISO in BD. 36 It is also important to note that many other markers are specific to particular disease states. For example, increased GPx levels were found in SCZ but not BD, 43 while protein carbonylation was elevated in BD but not in SCZ. 30 Overall, these findings support the hypothesis that mitochondrial dysfunction and oxidative imbalance contribute to the pathophysiology of BD and SCZ. Next, we explore upstream processes involved in controlling mitochondrial and ETC function in the context of BD and SCZ, including mitochondrial and nuclear genomics, mitochondrial dynamics and quality control, and Ca2+ homeostasis.
Mitochondrial and Nuclear Genomics
As BD and SCZ show a high degree of heritability, 45,46 studying where genetic mutations occur may provide insight into possible pathways that are dysregulated in these diseases. While genetic alterations related to countless cellular processes have been reported in BD and SCZ, 47,48 we focus on those directly related to ETC function. While mitochondria contain their own DNA, this is not sufficient for function of the ETC. 49 Both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA)–encoded proteins are essential for ETC assembly (Figure 1B). For example, complex I is composed of 37 nDNA-encoded and 7 mtDNA-encoded subunits. Importantly, a number of nDNA-encoded assembly factors or chaperones are required for the stability and proper assembly of ETC complexes. 50 nDNA-encoded proteins also regulate the replication, transcription, and repair of mtDNA. 51,52 Therefore, mutations in either nDNA or mtDNA have the potential to directly affect mitochondrial function.
mtDNA mutations likely contribute to BD and SCZ. mtDNA mutations (Table 1) are commonly associated with BD and SCZ. Furthermore, these disorders have higher rates of maternal inheritance than paternal inheritance, which aligns with the fact that mtDNA is inherited exclusively from the mother. 45,53 Dozens of mtDNA single-nucleotide polymorphisms (SNPs) in genes encoding ETC proteins have been associated with BD and SCZ. 54 –56 These SNPs have the potential to affect ETC function. For example, the mtDNA SNP 12027T>C, encoding the ND4 subunit of complex I, is associated with SCZ and with greater production of SOD, suggesting a compensatory response to increased ROS production. 57 mtDNA SNPs occurring in the ND1 and ND3 subunits of complex I are associated with BD and lead to impaired mitochondrial function in mtDNA cybrids. 54,58 Many other mtDNA SNPs have been associated with BD and SCZ, but the functional consequences of most of these have not been explored. Based on current evidence, it is likely that variants in genes encoding ETC proteins can directly affect mitochondrial function.
Mutations in nDNA are also involved in mitochondrial dysfunction. nDNA-encoded genes are important for the architecture, assembly, and catalytic functions of mitochondrial ETC. 49 Mutations in nDNA encoding complex I catalytic subunits have been shown to impair mitochondrial function. 59 Indeed, the complex I subunit NDUFV2 has been identified as a possible risk factor for BD; its gene is found at a well-replicated susceptibility locus for BD (18p11), and SNPs in this gene have been associated with BD. 60 One particular SNP (–602G>A) is associated with BD and results in decreased promoter activity. 61 Altered NDUFV2 expression levels have been reported in samples of both BD 60,62 and SCZ. 63 Downregulation of NDUFS7 has also been reported in BD, 30,31 which was correlated with reduced activity of complex I and increased protein oxidation. 30 nDNA-encoded proteins not only make up the majority of the ETC 64,65 but are also required for the stability of the ETC 50 and the replication, transcription, and repair of mtDNA. 51,52 For example, DNA polymerase subunit gamma (Polγ), which is responsible for the replication of mtDNA, is downregulated in peripheral cells of patients with BD. 66,67 Interestingly, transgenic mice expressing neuron-specific mutant Polγ accumulate mtDNA mutations and demonstrate mood disorder–like behavior that is worsened by tricyclic antidepressants and improved by Li+. 68 The association between genes and symptoms in psychiatric disorders is often complex, but identifying risk genes may help shed light on potential mechanisms and therapeutic targets for these disorders. 55,69,70
Mitochondrial Dynamics and Trafficking
Mitochondria are dynamic organelles that undergo changes in morphology and localization; these changes affect cellular processes such as ATP production, mitosis, and mitophagy. 71 –73 Mitophagy is a quality control process in which damaged mitochondria are degraded in lysosomes. Alterations in mitochondrial dynamics have been implicated in neurodegenerative diseases. 74 Initial investigations in psychiatric illnesses have suggested a role for altered mitochondrial dynamics in SCZ. However, these processes have not been thoroughly investigated in either SCZ or BD. Future research may help to answer these questions.
Mitochondrial dynamics involve changes in mitochondrial morphology through fusion and fission, as well as movement through the cells via microtubule motor proteins (Figure 1A). 71,75,76 Fusion promotes diffusion of contents between mitochondria, spreading metabolites and enzymes, 77 diluting damaged proteins and DNA, and increasing communications with the endoplasmic reticulum. 74,78 On the other hand, fission is necessary for cell division and for maintaining mitochondrial quality control; damaged mitochondria are isolated by fission and subject to mitophagy. 77 Thus, both fission and fusion are necessary for optimal mitochondrial and cell function.
The fusion process is mediated by Mitofusin 1 (Mfn1), Mitofusin 2 (Mfn2), 75,79 and optic atrophy 1 (Opa1) protein. 74 Mfn2 is also crucial for mitochondrial tethering to ER membranes, forming mitochondria-associated ER membranes (MAMs). 80 MAMs allow for the transport of Ca2+ and lipids from the ER to mitochondria and are important for mitophagy. 80,81 Knockout of Mfn2 in cells leads to fragmentation of mitochondria, disrupted MAMs, 80 and impaired mitophagy. 82 The role of fusion in psychiatric illnesses has not been established yet, but one analysis of postmortem prefrontal cortex samples revealed decreased levels of Opa1 in SCZ specimens. 83
Mitochondrial fission is essential for cell proliferation and mitophagy. 84,85 The main mediators of fission are mitochondrial fission 1 protein (Fis1) and dynamin-related protein 1 (Drp1). 74,86 However, few studies to date have established the role for Fis1 and Drp1 in BD and SCZ; these represent potential targets for future investigation. 83 An interesting mechanism for altered mitochondrial dynamics in SCZ is through G72. G72 has previously been identified as a candidate gene for SCZ, 87 and levels of G72 protein appear to be substantially higher in the plasma of both medicated and unmedicated patients with SCZ. 88 While originally thought to modulate NMDA signaling, a recent investigation demonstrated that transfection of G72 into primary neurons induces mitochondrial fragmentation and increases dendritic branching. 89 Cells expressing a loss-of-function Drp1 mutant, which have impaired fission, exhibit drastically reduced fragmentation in response to G72. 89 Overexpression of G72 may promote mitochondrial fission and lead to altered neural connectivity. G72 is not only a promising peripheral biomarker but may also contribute to mitochondrial dysfunction in SCZ.
Mitochondrial dynamics involve not only the processes of fission and fusion but also movement of mitochondria through the cell. 71 The trafficking and localization of mitochondria in neurons are influenced by ATP and Ca+2 concentration. 90 High levels of ATP increase mitochondrial motility in dendrites to areas of high energy demand 91 ; in contrast, high levels of ADP and Ca2+ inhibit movement. 91 –93 Because localization of mitochondria is important for Ca2+ homeostasis 10 and ATP provision, 94 impairment in mitochondrial trafficking can produce alterations in neurotransmitter release and synaptic function. 90,95 A key protein involved in mitochondrial trafficking is disrupted in schizophrenia (Disc1). 71,96 Disc1 associates with kinesin-1, which is necessary for the anterograde movement of mitochondria. 94 In yeast, decreased Disc1 function results in decreased complex I activity and ATP production, as well as perturbed Ca2+ buffering. 97 The Disc1 SNP R37 W decreases Disc1 expression 98 and impairs anterograde movement of mitochondria. 94 Other Disc1 SNPs are associated with SCZ and BD, although the functional consequences are unknown. 94,99,100 Currently available research suggests that mitochondrial dynamics are impaired in SCZ, but it is unknown if mitochondrial dynamics are involved in other psychiatric illnesses.
The Role of Calcium Homeostasis in Mitochondrial Dysfunction
Among other functions, Ca2+ influences cell metabolism, death/survival, and neurotransmission through regulation of the mitochondria. The primary uptake mechanism for Ca2+ by mitochondria is through the mitochondrial Ca2+ uniporter (MCU). 101,102 While the affinity of the MCU for Ca2+ is low, localization of mitochondria to regions of cytosolic Ca2+ influx, such as the ER and membrane channels, allows them to buffer the cell from large spikes in Ca2+ concentration. 10,102 Mitochondrial Ca2+ is extruded into the cytosol through exchangers that are coupled to the ETC (Figure 1 B). 9,102 Uptake of Ca2+ into mitochondria enhances respiration by activating several dehydrogenases in the citric acid cycle. 103 The subsequent accumulation of NADH leads to an increased production of ATP, which is required to pump Ca2+ out of mitochondria. 9,104 While Ca2+ is a positive effector of mitochondrial function, Ca2+ overload causes uncoupling of the ETC and depolarization of the mitochondrial membrane. 105,106 Ca2+ overload therefore decreases the mitochondrion’s capacity to generate ATP and remove Ca2+, 9 inducing ROS production 104 and potentially leading to apoptosis. As mitochondrial ATP production and Ca2+ buffering are essential for neurotransmission and cell survival, dysfunction may alter neural plasticity. 10,107
Intracellular Ca2+ dyshomeostasis has long been implicated in BD and SCZ. For example, elevated Ca2+ are frequently observed in stimulated platelets from patients with untreated SCZ and BD. 108 –112 Such findings are indicative of altered intracellular Ca2+ signaling but do not offer specific evidence of the processes involved. The ER is a likely source of Ca2+ dysfunction in BD and SCZ. Because the ER is a major source of intracellular Ca2+ and closely associated with mitochondria through MAMs, dysfunctions in ER-mediated release of Ca2+ affect Ca2+ homeostasis within mitochondria. 102,104
The ER releases Ca2+ largely through inositol triphosphate receptors (IP3 R). IP3 R is an ER membrane Ca2+ channel that is activated by IP3. Dysregulation of Ca2+ influx through IP3Rs disrupts mitochondrial function through Ca2+ overload. 104 In neurons, such events alter normal neurotransmitter release and synaptic plasticity. 10 Several lines of evidence suggest a role for IP3Rs in Ca2+ dyshomeostasis in BD and SCZ. Neuronal Ca2+ sensor-1 (NCS-1) protein levels are elevated in the dorsolateral PFC of patients with SCZ and BD but not depression. 113,114 NCS-1 increases cytosolic Ca2+ levels by enhancing IP3 R activity; this process is blocked by therapeutic levels of Li+. 115,116 Furthermore, Li+ and valproate inhibit enzymes in the IP cycle involved in the generation of IP3, decreasing IP3 R activity. 117,118 As well, both typical and atypical antipsychotics inhibit IP3-induced Ca2+ release. Patients with untreated BD or SCZ may have altered sensitivity to Ca2+-releasing stimuli in part due to overexpression of NCS-1. 119
The antiapoptotic protein Bcl-2 also inhibits IP3-mediated IP3 R Ca2+ release, 120 thereby protecting mitochondria against Ca2+ overload. 9 Decreased levels of Bcl-2 protein have been reported in the frontal cortex of patients with BD 121 and the temporal cortex of patients with SCZ. 122,123 Bcl-2 SNPs in patients with BD are associated with lower levels of Bcl-2 messenger RNA and protein, elevated basal Ca2+, and enhanced IP3R-mediated Ca2+ release. 124,125 Li+ has been shown to increase Bcl-2 expression in the central nervous system 126 and restores Ca2+ homeostasis in Bcl-2 variants, further supporting a role for Bcl-2 in BD. 124 Bcl-2 not only reduces IP3R-mediated Ca2+ release 120 but also modulates membrane L-type Ca2+ channels. 127 Cells with decreased levels of Bcl-2, such as in BD and SCZ, are therefore at greater risk of mitochondrial Ca2+ overload. Ca2+ dysregulation observed in BD and SCZ may be in part due to altered IP3 R activity, leading to enhanced influx of Ca2+ from the ER to closely associated mitochondria. Ca2+ overload can depolarize the mitochondrial membrane and impair its other functions. 105,106
It is important also to consider that mitochondrial function is crucial in the regulation of intracellular Ca2+ levels. Impairments in mitochondrial function have the potential to dysregulate Ca2+ homeostasis. For example, the mtDNA SNP 10398G>A, found in the region encoding the complex I subunit ND3, is associated with BD 54 and with higher mitochondrial pH and Ca2+ concentrations. 128 Moreover, SH-SY5Y cells chronically exposed to rotenone (>2 weeks), which induces 15% to 30% decreases in complex I function, demonstrate altered Ca2+ influx in response to stimulation, which is dependent in part on MCU. 129 This suggests that diminished ETC function, which is commonly observed in BD and SCZ, impairs the mitochondrion’s ability to buffer intracellular Ca2+. Mitochondrial ROS can also alter Ca2+ flux by modulating redox-sensitive Ca2+ channels, such as TRPM2. 130,131 Disruptions in either mitochondrial function or intracellular Ca2+ homeostasis have the potential to exacerbate each other.
Concluding Remarks
We have described a role for mitochondrial dysfunction in the pathophysiology of major psychoses and discussed upstream pathways that may contribute to controlling mitochondrial function in BD and SCZ. Upstream pathways can be summarized as mutations in mtDNA and nDNA, perturbed mitochondrial dynamics, and dysregulated intracellular Ca2+ homeostasis. BD and SCZ are complex and heterogeneous diseases; it is unlikely that all of these pathways are dysregulated in a given individual. Rather, different individuals may present with alterations in different processes that may converge upon mitochondrial dysfunction. The processes involved in energy production, mitochondrial dynamics, and Ca2+ homeostasis are interdependent. Subtle alterations in one process may either be compensated or exacerbated by other processes, contributing to the complexity of these disorders. Despite this, the mitochondrial dysfunction present in BD appears to be distinct from that in SCZ. While both BD and SCZ are associated with mitochondrial dysfunction and redox modulations, BD is associated with decreased protein and gene expression of NDUFS7 and NDUFS8, 2 core subunits for electron transfer in complex I. As these subunits are mandatory for electron transfer to ubiquinone, patients with BD may be more susceptible to ROS generation by electron loss through complex I compared to those with SCZ. Altered expression of complex I subunits is reported in SCZ, but the direction of these findings is inconsistent, unlike the decreased levels observed in BD. 4
BD features manic and depressive episodes. Studies to date have not been designed to address how mitochondrial function may change in each state of the illness within the same individual. Nevertheless, we might speculate that during manic episodes, patients experience a general increase in neurotransmission; this requires high levels of energy, suggesting an increase in mitochondria activity resulting in increased ATP and ROS production. Overproduction of mitochondrial ROS leads to changes in the redox state of certain proteins, altering their function. This may explain, in part, the cyclical nature of BD. Redox modulations to proteins may decrease mitochondrial activity, altering neurotransmission and producing symptoms of depression. Of course, it should be noted that the above mechanism is speculative, and longitudinal studies examining markers of mitochondrial function are crucial to determine how mitochondrial activity varies between mood states.
Understanding the upstream processes that affect mitochondrial function will help in identifying the different triggers of mitochondrial dysfunction in each of the 2 psychiatric illnesses. Ultimately, delineating the causes of mitochondrial dysfunction will guide rational development of novel therapeutics with better efficacy and fewer adverse effects.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We thank the Brazilian government (CAPES) for supporting AM for his exchange doctorate and TMS in an exchange fellowship (CNPq- SWB). ACA is funded by CHIR (MOP-143439), Ontario Mental Health Foundation, and Ontario Ministry of Research and Innovation (ERA-14-10-022).