
Abstract
Objectives
Salidroside is used for treating inflammation-based diseases; however, its molecular mechanism is unclear. In this study, we determined the protective role of salidroside on the endotoxin-induced damage caused to the mouse alveolar epithelial type II (MLE-12) cells and its underlying mechanism.
Methods
An in vitro model for acute lung injury was constructed by inducing the MLE-12 cells using lipopolysaccharide (lipopolysaccharides, 1 mg/L). Then, The MTT assay was conducted to assess the survival rate of the MLE-12 cells in the different groups. After the treatment, apoptosis of MLE-12 cells was determined, and the mRNA and protein expression of miR-199a-5p, HMGB1, NF-kB65, TNFAIP8L2, p-IkB-α, and TLR4 was estimated by Western Blotting and RT-PCR. ELISA was also used to measure the concentration of inflammatory cytokine molecules IL-1β, IL-6, TNF-α, and IL-18 in the cell-free supernatant. Lastly, cell morphology was examined using the AO/EB technique.
Results
We showed that salidroside reduced the protein and gene expression of HMGB1, NF-kB65, miR-199a-5p, p-IkB-α, and TLR4, whereas it increased the gene and protein expression of TNFAIP8L2. Furthermore, it decreased the concentrations of cytokine molecules like IL-1β, IL-6, TNF-α, and IL-18 in the cell-free supernatant. MLE-12 also showed a lower apoptosis rate, higher survival rate, and better cell morphology.
Conclusion
Salidroside significantly inhibited the LPS-induced MLE-12 cell damage. Our results suggest that this could be by reducing miR-199a-5p and enhancing TNFAIP8L2 expression.
Keywords
Introduction
Sepsis is a deadly immune disorder induced by systemic inflammatory response syndrome (SIRS) triggered by a severe infection, which can cause multi-organ failure. Lungs are one of the most easily affected organs by sepsis, where ≈50% of the sepsis-affected patients develop acute respiratory distress syndrome (ARDS).1,2 Due to ARDS, these patients further develop severe hypoxia, which leads to organ failure, and can be fatal. 3 Alveolar epithelial cell damage is the primary pathogenesis of sepsis ARDS. 4 Recent studies indicate that the inflammatory response activated by severe infection is primarily responsible for acute lung damage. Regulating this inflammatory response could help decrease sepsis-induced acute lung injury.
Tumor necrosis factor-inducible protein 8-like protein 2 (TNFAIP8L2) is a novel negative regulator of the body’s innate immune response. 5 It is expressed in various cells and plays a vital role in the inflammatory response. 5 Lipopolysaccharides (LPS) can activate macrophages in cells with TNFAIP8L2 gene deletion and increase the TNF-α and IL-6 levels compared to normal macrophages. Similarly, protein and gene expressions of IL-6 were significantly upregulated when the RAW264.7 macrophage cell line with a suppressed TNFAIP8L2 expression was induced by LPS in vitro. 6
MicroRNA (miRNA) belongs to a set of endogenous small single-stranded non-coding RNA (ncRNA). miRNAs are typically 18–24 nucleotides long and are involved in several biological processes, such as inflammation, apoptosis, oxidative stress, tumorigenesis, cell proliferation, tissue growth, and organ formation.7–9 miRNA binds wholly or partially to the mRNA’s 3′-untranslated region (3′-UTR), thereby degrading or blocking the mRNA expression of the gene of interest. Studies have linked miRNAs to the onset and progression of acute lung injury, lung cancer, pulmonary fibrosis, and pulmonary tuberculosis.10,11 It was observed that in sepsis, miR-199a-5p suppressed the expression of the surfactant protein D and activated the NF-κB pathway, leading to the failure of the intestinal barrier. 12 This led to the hypothesis that miR-199a-5p played a vital role in the onset and progression of sepsis-induced acute pulmonary damage; however, the actual molecular mechanism is still unknown.
Salidroside is a primary active molecule extracted from Salidrosea, which shows anti-tumor activity in bladder, breast, and colon cancer. 13 Studies suggest that salidroside suppresses the expression of the high glucose-induced matrix metalloproteinases and inflammatory responses in the endothelial cells by inhibiting the expression of ROS-related HMGB1-TLR4 signaling. 14 Furthermore, salidroside inhibits the NLRP3 activity, thereby decreasing the inflammatory response of the non-alcoholic fatty liver cells and improving the fat metabolism of liver cells. 15 It also upregulates SIRT1 and inhibits the NF-κB and HMGB1 pathways to reduce the effects of sepsis and lung injuries caused due to a mechanical ventilator.16,17 It triggers the lung epithelial cells to secrete miRNA-146a exosomes, which helps in regulating the inflammatory pathway of alveolar macrophages. 18 Salidroside has also been shown to regulate the sepsis-induced pulmonary inflammatory response; 19 however, the exact mechanism is still unclear. No reports have been published regarding the protective mechanism of salidroside that helps regulate miR-199a-5p/TNFAIP8L2 against the endotoxin-induced mouse alveolar epithelial type II (MLE-12) cells. Here, using TargetScans, we found that miR-199a-5p targeted the regulation of TNFAIP8L2. Salidroside improved TNFAIP8L2 expression, inhibited the sepsis-induced inflammation and repaired the sepsis-induced acute lung injuries by inhibiting the miR-199a-5p expression. We also showed that the role of salidroside in septic-induced acute lung damage was by regulating the miR-199a-5p/TNFAIP8L2 pathway. Our results presented a novel therapeutic agent and described the probable mechanism used by salidroside, which could offer insights while treating sepsis-induced acute lung injuries.
Materials and methods
Drugs
UM-164 (a TNFAIP8L2 inhibitor, CAS: 903564-48-7, purity ≥98%) was obtained from Shanghai Loulan Biotechnology Co., Ltd. (Shanghai, China). 20 Salidroside (CAS: 10338-51-9, purity ≥98%) was obtained from Chengdu Refensi Biotechnology Co., Ltd. (Chengdu, China).
MLE-12 cell culture and grouping
The Mouse Lung Epithelial-12 (MLE-12) cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Cat. No. FS-X0413, Shanghai, China) and cultured in the RPMI-1640 cell medium (Cat. No.PM150110B, Wuhan BOSTER Biotechnology Co., Ltd., China) containing 15% v/v Fetal Bovine Serum (FBS). Cells were cultured in an incubator with 5% CO2 at a saturated humidity of 37°C. Cells were sub-cultured when the growth density was 80%. After confluence reached ≈60%, the cells (1 × 105 cells/mL) were transferred into 25 cm2 culture bottles and cultivated synchronously for 24 h. To study the effect of LPS on the MLE-12 cell viability, cells were cultured in varying LPS concentrations (such as 0.5, 1.0, 1.5, and 2.0 mg/L), allowing them to grow for 24 h. The effect of salidroside on the LPS-induced cell damage was determined after categorizing the cells into various groups, that is, normal control group, LPS-induced group, LPS+varying concentrations of salidroside (25–75 μmol/L) groups. The cells in these groups were cultured for 24 h and then tested.
Cytotoxicity test
CCK-8 technique was implemented for testing cytotoxicity. MLE-12 cells (1 × 103 cell density) were inoculated into every well of the 96-well culture plates. Different salidroside concentrations (0, 25, 50, 75, 100, and 150 μmol/L) were pipetted into the respective test groups, and every experimental group was assigned 6 repeats. All wells were cultured for 48 h. Subsequently, the CCK-8 solution (10 μL, Cat. No. ab228554, Cambridge, UK) was added to the culture medium, and its absorbance was measured at 450 nm wavelength using the microplate reader after 4 h.
MLE-12 cells transfection
The log-phase MLE-12 cells (1 × 105 cells/mL) were inoculated into the 6-well culture plates. When the confluence of the cells increased to 60%, the miR-199a-5p mimic, mimic control, miR-199a-5p inhibitor, and inhibitor control to the cells were transfected with a concentration of 50 nmol/L, as per the instructions provided with the Lipofectamine 2000 transfection reagent. After culturing the samples for 6 h, the culture medium was replaced, a fresh medium was added, and the cells were cultured for another 24 h. Subsequently, the samples were collected for RT-PCR and Western blotting analyses.
Determining the correlation between miR-199a-5p and TNFAIP8L2
The TargetScan bioinformatics software was used to determine the presence of continuous binding sites in the 3′UTR of TNFAIP8L2 and the miR-199a-5p nucleotide sequence. TNFAIP8L2 wild-type (TNFAIP8L2-WT) and the mutant double luciferase reporter plasmid (TNFAIP8L2-MUT) were constructed. Thereafter, miR-199a-5p control and TNFAIP8L2-WT (miR-NC+ TNFAIP8L2-WT), miR-199a-5p mimic and TNFAIP8L2-WT (miR-199a-5p+TNFAIP8L2-WT), miR-199a-5p mimic and TNFAIP8L2-MUT (miR-199a-5p+TNFAIP8L2-MUT), and the miR-199a-5p control and TNFAIP8L2-MUT (miR-NC+TNFAIP8L2-MUT) were co-transfected into the cells, respectively. The different test cells were cultured for 48 h, and their luciferase activity was measured according to the manufacturer’s instructions. Then, the cells were inoculated from the miR-199a-5p inhibitor-NC, Mir-199a-5p inhibitor, miR-199a-5p mimic-NC, and the miR-199a-5p mimic groups into the 24-well cell culture plates, and were incubated for 24 h. Finally, the TNFAIP8L2 protein expression was detected using the Western Blot test.
Real-time quantitative reverse transcription polymerase chain reaction
Primer sequences of RT-PCR analysis.

Western blot
MLE-12 cells were treated with LPS and/or salidroside for 24 h. The total protein of the cells was then extracted using the radioimmunoprecipitation assay (RIPA). The cytoplasmic and nuclear protein kit (Applied Biosystems, CA, USA) was used for extracting the nucleoproteins. The total protein concentration was estimated using the BCA kit, and a small quantity of the total protein (30 μg) was extracted. Western Blot was used to detect the target protein’s expression. After the proteins were separated on the Sodium Dodecyl Sulfate-PolyAcrylamide Gel Electrophoresis (SDS-PAGE), the gel was transferred to a Polyvinylidene fluoride (PVDF) membrane at the standard conditions, that is, Voltage of 100 V, at room temperature for 60 min. Subsequently, the membrane was placed in TBST (Tris Buffered Saline with 0.1% Tween) + skim milk powder [5% (w/v)] solution, sealed, and incubated for 60 min at room temperature. After incubation, the membrane was washed three times using TBST for 5 min each. The membrane was then incubated with the diluted primary antibodies for the β-actin (Cat. No. ab179467, Abcam, Cambridge, UK), p-IκB-α (Cat. no. 2859, Cell Signaling Technology, Inc., Danvers, MA, USA), NF-κB65 (Cat. No.90479, Shanghai Jingke Chemical Technology Co., Ltd., Shanghai, China), IκB-α (Cat. No. sc-74451 Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), TLR4 (Cat. No. ab22048, Abcam, Cambridge, UK), TNFAIP8L2 (Cat. No. ab16916, Abcam, Cambridge, UK), and HMGB1 (Cat. No. ab77302, Abcam, Cambridge, UK) proteins, at 4°C, overnight. After incubation, the membrane was washed thrice using TBST for 5 min each. Subsequently, it was incubated with the respective secondary antibodies rabbit anti-mouse IgG H&L (HRP, Cat. No. ab6728, Abcam, Cambridge, UK) for an hour at 37°C. The membrane was washed with TBST, as mentioned above. Finally, the Enhanced Chemiluminescence (ECL) chromogenic solution was added and incubated with the film for 2 min at room temperature. Then, the target protein bands were analyzed using ImageJ software (NIH, USA). β-actin was used at a dilution of 1:5000, while other antibodies were diluted to 1:1000.
ELISA assay
A double antibody sandwich ELISA technique was used in this study. The cell-free supernatant of the LPS-induced MLE-12 cells, with or without salidroside treatment, was collected to estimate the cytokine concentration using the commercial kits (R&D Systems, Inc. Minneapolis, MN, USA) based on the manufacturer’s instructions. Each sample was tested in triplicates, and the result was expressed as the mean.
Flow cytometry
The MLE-12 cells were cultured in each group for 24 h, washed twice using the Phosphate Buffer Saline (PBS), and then suspended into the binding buffer. Annexin V-FITC (10 μL) was added to the cells, followed by propidium iodide (PI) (5 μL), and the mixture was incubated in the dark, at room temperature, for 15 min. The flow cytometer was then used to detect cell apoptosis.
Immunofluorescence
MLE-12 cells were cultured in 24-well culture plates. After 60% of the cells had adhered to the wells, the cells were stimulated using a drug for 24 h. The adhered cells were then washed with PBS to remove all traces of the drug and fixed with paraformaldehyde (v/v, 4%) for 30 min at room temperature. The cells were permeabilized using Triton X-100 (v/v, 0.5%) for 15 min at room temperature, sealed, and incubated at room temperature for 90 min. The MLE-12 cells were incubated with a primary antibody (Ki67 1:400, Cat. No. ab279653, Abcam, Cambridge, UK) overnight at 4°C. After incubation, the cells were washed with PBS to remove the excess antibody and again incubated with a fluorescent secondary antibody (1:400 dilution, Sigma, St. Louis, MO, USA) at room temperature, in the dark, for an hour. DAPI staining was used to stain the DNA. The localization and distribution of proteins within the cells were detected with a fluorescence microscope (YJ-2002H, Shanghai Gongzhou Valve Co., Ltd. Shanghai, China).
Hoechst technique
The log-phase MLE-12 cells (3 mL, 5 × 107/L) were inoculated into the 6-well culture plates. The cells were cultured in the incubator and allowed to adhere to the walls of the culture plates. LPS (1.0 mg/L) and salidroside (25, 50, and 75 μmol/L) were added to these cultured cells and further incubated for 24 h. The cells were then with PBS to remove the traces of the added molecules. Finally, 1 mL of PBS was added to each well. Subsequently, paraformaldehyde (4% v/v) was added to each well and stained as per the instructions in the Hoechst 33,258 apoptosis kit (Cat. No. 9754, Beijing Biolab Technology Co., Ltd., Beijing, China). The stained cells were observed and imaged using a fluorescence microscope.
Statistical analysis
Statistical analyses of all experimental data were conducted using the SPSS ver. 22.0 software. The normally distributed data were presented as the mean ± standard deviation (SD). The mean value represented the average of 3 independent sets of experiments. Independent sample T-tests and the One-Way ANOVA tests compared the data from different groups. Finally, the Newman–Keuls (SNK)-q test was used for additional pairwise comparison.
Results
The miR-199a-5p inhibitor affects the viability and the inflammatory mediatory response of the LPS-induced MLE-12 cells
We analyzed the impact of the miR-199a-5p inhibitor on the activity and inflammatory response of the LPS-induced MLE-12 cells. The miR-199a-5p inhibitor was transfected into the MLE-12 cells, and the test cells were cultured with 1 mg/L of LPS for 24 h. We determined the viability of the MLE-12 cells using the CCK-8 assay and measured the concentration of the inflammatory cytokines like IL-6, IL-1β, TNF-α, and IL-18 in the cell-free supernatant using the ELISA technique. We found that miR-199a-5p inhibitor significantly decreased the concentration of the IL-6, IL-1β, TNF-α, and IL-18 in the cell-free supernatant, while their viability was increased in the LPS-induced group (Figure 1(a)–(e)). Effect of the miR-199a-5p inhibitor on the activity of TNFAIP8L2/TLR4/NF-kB axis, viability, and inflammation response of the LPS-treated MLE-12 cells. The MLE-12 cells were transfected with the miR-199a-5p inhibitor, and the test cells were additionally cultured in the presence of LPS (1 mg/L) for 24 h. The MLE-12 cellular viability was measured using the CCK-8 technique, while the levels of the IL-6, IL-1β, TNF-α, and IL-18 molecules in the cell-free supernatant were measured using ELISA. The protein expression of p-IκB-α, NF-κB65, TLR4, NFAIP8L2, and HMGB1 was measured using the Western Blotting technique. miR-199a-5p gene expression was determined with the RT-PCR. The data in the study are depicted as Mean ± SD. **p < .05 when compared to the control. Control: control group; Inhibitor control: miR-199a-5p inhibitor control group; Inhibitor: miR-199a-5p inhibitor group; Model: model group.
The miR-199a-5p inhibitor affects the protein expression of TNFAIP8L2/TLR4/NF-kB in the LPS-treated MLE-12 cells
To observe the effect of miR-199a-5p on the protein expression of TNFAIP8L2/TLR4/NF-kB in the LPS-treated MLE-12 cells, we transfected the MLE-12 cells with the miR-199a-5p inhibitor and cultured them with 1 mg/L LPS for 24 h. We used the Western blot to determine the expression of NF-κB65, p-IκB-α, TNFAIP8L2, TLR4, and HMGB1 proteins. We also assessed miR-199a-5p gene expression using RT-PCR. We found a reduced expression of miR-199a-5p in the transfected cells. Additionally, there was a decrease in the expression of various proteins like p-IκB-α, NF-κB65, TLR4, and HMGB1, while the TNFAIP8L2 expression was elevated (Figure 1(f)–(k)). Thus, our results suggested that miR-199a-5p regulates the inflammation-mediated response in the MLE-12 cells by regulating the activity and expression of the molecules in the TNFAIP8L2/TLR4/NF-kB pathway.
miR-199a-5p inhibitor combined with UM-164 affects the cell viability, inflammation, and activity of the TNFAIP8L2/TLR4/NF-kB pathway in the LPS-induced MLE-12 cells
UM-164, a TNFAIP8L2 inhibitor, can significantly inhibit the expression of TNFAIP8L2 in inflammatory tissues and cells.
20
To analyze the combined impact of the miR-199a-5p inhibitor + UM-164 on inflammation, we examined the expression levels and activity of the TNFAIP8L2/TLR4/NF-kB pathway in the LPS-induced MLE-12 cells. We transfected the miR-199a-5p inhibitor into MLE-12 cells. LPS (1.0 mg/L) and UM-164 (1 μM) were then added to the cell culture and incubated for 24 h. We evaluated the expression of NF-κB65, p-IκB-α, TLR4, TNFAIP8L2, and HMGB1 using western blotting. The levels of the inflammatory cytokines like IL-6, IL-1β, TNF-α, and IL-18 in the cell-free supernatant were measured using the ELISA technique. The results showed that the miR-199a-5p inhibitor increased the TNFAIP8L2 expression (Figure 2(a) and (b)) and markedly decreased the expression of the p-IκB-α, NF-κB65, TLR4, and HMGB1 proteins in the MLE-12 cells (Figure 2(a)–(f)). The combined impact of the miR-199a-5p inhibitor + UM-164 on cell viability, inflammation, and activity of the TNFAIP8L2/TLR4/NF-kB pathway in the LPS-induced MLE-12 cells. MLE-12 cells were transfected using the miR-199a-5p inhibitor, and the cells in the test groups were additionally cultured in the presence of 1 mg/L of LPS and 1 μM of UM-164 for 24 h. The expression of NF-κB65, p-IκB-α, TLR4, NFAIP8L2, and HMGB1 proteins were determined using Western blotting. The MLE-12 cellular viability was measured using the CCK-8 technique, while the levels of the IL-6, IL-1β, TNF-α, and IL-18 molecules in the cell-free supernatant were measured using ELISA. The data were presented as Mean ± SD. **p < .05 when compared to the control. Inhibitor: miR-199a-5p inhibitor group.
However, UM-164 significantly decreased the TNFAIP8L2 expression and increased the expression of the p-IκB-α, NF-κB65, TLR4, and HMGB1 proteins (Figure 2(a)–(f)), and increased the levels of the inflammatory cytokines IL-6, IL-1β, TNF-α, and IL-18 in LPS-induced MLE-12 cells (Figure 2(g)–(k)). Compared with the UM-164 group, miR-199a-5p inhibitor combined with UM-164 markedly increased the TNFAIP8L2 expression (Figure 2(a) and (b)), markedly decreased the expression of the p-IκB-α, NF-κB65, TLR4, and HMGB1 proteins (Figure 2(a)–(f)), and reduced the levels of the inflammatory cytokines IL-6, IL-1β, TNF-α, and IL-18 in LPS-induced MLE-12 cells (Figure 2(g)–(k)). The results further stated that the TNFAIP8L2 attenuates the effect of miR-199a-5p on LPS-induced MLE-12 cells inflammation.
TNFAIP8L2 acts like a regulatory target of miR-199a-5p
To analyze whether miR-199a-5p could modulate LPS-induced acute lung injury by regulating TNFAIP8L2, we used the TargetScan software to derive the prediction results and found that the 3′UTR sequence of TNFAIP8L2 included the nucleotide sequence which complemented that of miR-199a-5p (Figure 3(d)). We then compared the relative luciferase activities of different groups and found that the wild-type WT-TNFAIP8L2 activity in the miR-199a-5p mimic group was significantly reduced than in the miR-NC group (Figure 3(e)). Furthermore, the relative luciferase activity in the MUT-TNFAIP8L2 was not significantly altered (Figure 3(e)). However, the TNFAIP8L2 expression in the miR-199a-5p mimic group was significantly decreased than in the miR-199a-5p mimic-NC group (Figure 3(a)–(c)). We further showed that the miR-199a-5p inhibitor group showed a higher TNFAIP8L2 expression than the miR-199a-5p inhibitor-NC group (Figure 3(a)–(c)). Together, our results indicate an antagonistic relationship between miR-199a-5p and TNFAIP8L2. TNFAIP8L2 is the regulatory target of miR-199a-5p. (a) MiR-199a-5p mimic group cells inhibited the TNFAIP8L2 gene expression in LPS-treated MLE-12 cells. 
Salidroside affects the TNFAIP8L2/TLR4/NF-kB pathway and miR-199a-5p expression in the LPS-induced MLE-12 cells
To determine the influence of salidroside on the TNFAIP8L2/TLR4/NF-kB pathway and miR-199a-5p expression in the LPS-treated MLE-12 cells, we cultured the MLE-12 cells in the presence of 1 mg/L of LPS and varying concentrations of salidroside (25–75 μmol/L) for 24 h. Protein expression levels of the TNFAIP8L2, p-IκB-α, NF-κB65, TLR4, and HMGB1 were determined using Western Blotting, and RT-PCR was used for determining the mRNA expression of miR-199a-5p TNFAIP8L2, IκB-α, NF-κB65, TLR4, and HMGB1. Salidroside treatment significantly reduced the miR-199a-5p, TLR4, NF-κB65, and HMGB1 mRNA expression (Figure 4(g)–(l)) and p-IκB-α, NF-κB65, TLR4, and HMGB1 protein expression (Figure 4(a)–(f)). However, it increased the TNFAIP8L2 mRNA and protein expression levels (Figure 4(d) and (j)). The results indicated that salidroside inhibited the activities of the molecules involved in the inflammatory pathways in LPS-induced MLE-12 cells, possibly by decreasing miR-199a-5p and enhancing TNFAIP8L2 expression. Salidroside affects the TNFAIP8L2/TLR4/NF-kB pathway and miR-199a-5p expression in the LPS-induced MLE-12 cells. The MLE-12 cells were cultured in the presence of LPS (1 mg/L) and varying concentrations of salidroside (25–75 μmol/L) for 24 h. The protein expression levels of the NFAIP8L2, p-IκB-α, NF-κB65, TLR4, and HMGB1 were determined using the Western Blotting technique, while the mRNA expression of miR-199a-5p, NFAIP8L2, IκB-α, NF-κB65, TLR4, and HMGB1 was determined using the RT-PCR technique. The data is depicted as Mean ± SD. **p < .05, when compared to control. Control: control group; Model: model group.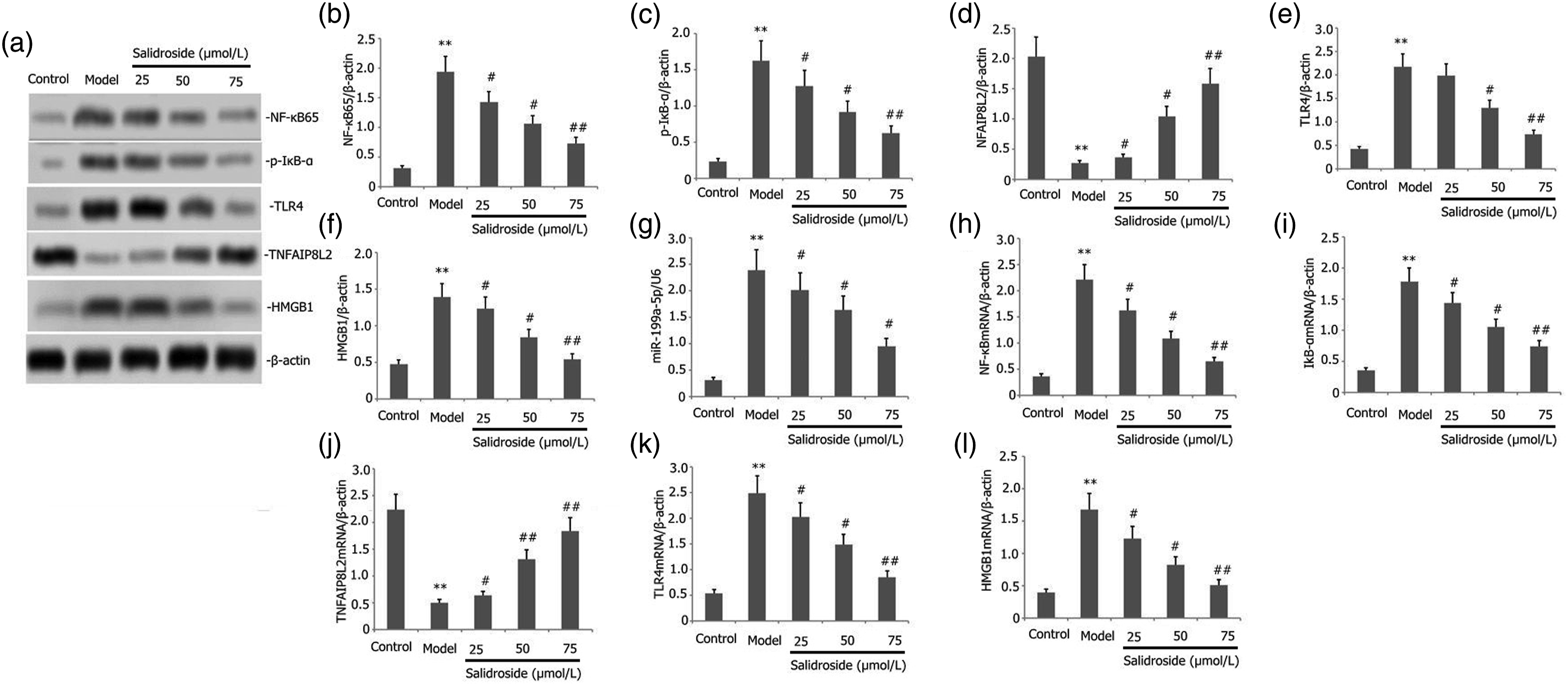
Salidroside affects the inflammatory mediators in the LPS-induced MLE-12 cells
To determine the influence of salidroside on the inflammation mediating molecules, we cultured the MLE-12 cells in the presence of 1 mg/L of LPS and varying concentrations of salidroside (25–75 μmol/L) for 24 h. We determined the IL-1β, IL-6, TNF-α, and IL-18 levels in the cell-free supernatant using ELISA. Salidroside significantly decreased the concentration of IL-1β, IL-6, TNF-α, and IL-18 in the cell-free supernatant (Figure 5(b)–(e)). The results indicated that salidroside reduced the inflammation of the LPS-induced MLE-12 cells. Salidroside affected the viability, inflammation, and apoptosis of the LPS-induced MLE-12 cells. The MLE-12 cells were cultured in the presence of LPS (1 mg/L) and varying concentrations of salidroside (25–75 μmol/L) for 24 h. The viability of the MLE-12 cells was estimated using the CCK-8 technique, and the morphology and apoptosis of the cells were assessed using the Hoechst assay. The IL-1β, IL-6, TNF-α, and IL-18 levels were determined in the cell-free supernatant using ELISA. The data is depicted as Mean ± SD. **p < .05 when compared to the control. Control: control group; Model: model group.
Salidroside affects viability, cell morphology, and apoptosis of the LPS-treated MLE-12 cells
To determine the influence of salidroside on the viability, cell morphology, and apoptosis, we cultured the cells in 1 mg/L of LPS and varying concentrations of salidroside (25–75 μmol/L) for 24 h. We then estimated the viability of the MLE-12 cells using the CCK-8 technique and assessed the morphology and apoptosis of the MLE-12 cells using the Hoechst assay. Results showed that the salidroside treatment ameliorated cell morphology and apoptosis of the LPS-treated MLE-12 cells and enhanced their viability (Figure 5(a), (f) and (g)).
The combined influence of salidroside + miR-199a-5p mimics the TNFAIP8L2/TLR4/NF-KB pathway
The researchers transfected the miR-199a-5p mimic sequence into the MLE-12 cells grown in the presence of 1 mg/L of LPS and salidroside (50 μM) for 24 h. We then determined the protein expression of p-IκB-α, NF-κB65, TLR4, NFAIP8L2, and HMGB1 proteins in the MLE-12 cells using Western blotting. We found that the miR-199a-5p mimic significantly inhibited TNFAIP8L2 expression and enhanced the expression of TLR4, NF-κB65, p-IκB-α, and HMGB1 (Figure 6(a)–(f)). However, salidroside inhibited the miR-199a-5p expression, reduced the expression of the TLR4, NF-κB65, p-IκB-α, and HMGB1 proteins, and increased the TNFAIP8L2 expression in MLE-12 cells transfected with miR-199a-5p mimic. The combined influence of salidroside + miR-199a-5p mimic on the TNFAIP8L2/TLR4/NF-KB pathway. The MiR-199a-5p mimic was transfected into the MLE-12 cells and grown in the presence of 1 mg/L of LPS and 50 μM of salidroside for 24 h. The protein expression of the p-IκB-α, NF-κB65, NFAIP8L2, TLR4, and HMGB1 proteins in the MLE-12 cells was then determined using western blotting. The data are depicted as Mean ± SD. **p < .05, when compared to control. Mimic: miR-199a-5p mimic group.
The combined effect of salidroside + miR-199a-5p mimic on inflammation, viability, morphology, and apoptosis of the MLE-12 cells
We transfected the miR-199a-5p mimic into the MLE-12 cells that were grown in the presence of 1 mg/L of LPS and 50 μM of salidroside for 24 h. We then determined the viability of the MLE-12 cells using the CCK-8 technique. At the same time, the apoptosis and cell morphology of the MLE-12 cells were assessed using the Hoechst and TUNEL techniques, respectively. The IL-1β, IL-6, TNF-α, and IL-18 levels in the cell-free supernatant were determined using ELISA. We found that the inclusion of the miR-199a-5p mimic sequence enhanced the IL-1β, IL-6, TNF-α, and IL-18 levels and the apoptotic activity of the LPS-induced MLE-12 cells, reduced the MLE-12 cell viability and increased their damage (Figure 7(a)–(g)). When combined with salidroside, the miR-199a-5p mimic decreased the IL-1β, IL-6, TNF-α, and IL-18 levels, and the apoptotic activity of the LPS-induced MLE-12 cells, enhanced the MLE-12 cell viability and helped in healing the injuries caused to the LPS-induced MLE-12 cells (Figure 7(a)–(g)). Hoechst 33,258 fluorescence staining results indicated that the control cells had a uniform shape and size, with a clear boundary, and emitted a weak normal blue fluorescence, with no observable morphological changes in the nucleus of the cells (Figure 7(h)). However, after the LPS treatment, all cells in the various test groups displayed a strong and bright fluorescence with a dense dye under the fluorescent microscope. (Figure 7(h)). Furthermore, when we transfected the cells with a miR-199a-5p mimic, we found a significant rise in densely stained cells, suggesting that apoptosis is increased (Figure 7(h)). However, the salidroside treatment reduced the number and the apoptosis of the densely stained cells. The combined influence of salidroside + miR-199a-5p mimic on the expression, inflammation, and apoptosis of the MLE-12 cells. The MiR-199a-5p mimic was transfected into the MLE-12 cells and grown in the presence of 1 mg/L of LPS and 50 μM of salidroside for 24. The viability of the MLE-12 cells was estimated using the CCK-8 technique, while the apoptosis and cell morphology of the MLE-12 cells was evaluated using the Hoechst techniques. The IL-6, IL-1β, TNF-α, and IL-18 levels in the cell-free supernatant were determined using the ELISA. The data are depicted as Mean ± SD. **p < .05, when compared to control. Mimic: miR-199a-5p mimic group.
Discussion
Acute lung damage occurs due to the uncontrolled pulmonary inflammatory response to multiple intrapulmonary/extrapulmonary causes. A pulmonary inflammatory syndrome is characterized by an imbalance of local or systemic inflammatory responses that increases alveolar-capillary membrane permeability. 21 Due to the rapid progression of the disease and the lack of effective treatment, its case fatality rate remains high, making it a hotspot and a challenge in clinical medical research. However, its specific pathogenesis has not been fully elucidated. Many studies have shown that the endotoxins and lipopolysaccharides produced by the gram-negative bacteria mediate the generation and release of several cytokines and inflammatory mediators by the inflammatory cells (like mononuclear macrophages, neutrophils, and endothelial cells), affecting the pathogenesis of the acute lung injuries.22,23 Therefore, regulating endotoxin and LPS-induced inflammatory response will help manage the acute lung injury inflammatory response and reduce lung injury. In this study, salidroside reduced inflammation and damage to the LPS-treated MLE-12 cells and assuaged the resulting acute lung injury.
Studies have shown that the TNFAIP8L2 knockout mice eventually develop leukocytosis, splenomegaly, weight loss, multi-organ failure, and produce inflammatory mediators spontaneously, 24 after the LPS attack, sepsis and even premature death occur due to excessive inflammatory response, and serological tests of the animals revealed an elevated expression of IL-1, TNF-α, IL-10, IL-6, and IL-12 levels. 24 Furthermore, there was a significant rise in the number of T cells, B cells, and dendritic cells. 24 Recent studies stated that TNFAIP8L2 inhibited the generation of the pro-inflammatory mediating molecules by regulating the pro-inflammatory HMGB1/TLR4/NF-κB signaling pathways. 25 During sepsis, a protein kinase phosphorylates and degrades the NF-κB inhibitor, IκB, from its trimer complex. This transfers the IκB from the cytoplasm to the cell’s nucleus. NF-κB then interacts with its binding site and initiates the transcription and translation of numerous cytokine genes (IL-1, TNF-α, and IL-6), activating the inflammatory cells. 26 The HMGB1/Toll-like receptor 4 (TLR4)/nuclear factor-κB (NF-κB) signaling pathway secretes many vital molecules in the inflammatory signal transduction pathway. 27 Here, we treated the MLE-12 cells with LPS, which inhibited the expression of TNFAIP8L2 and reduced the HMGB1/TLR4/NF-κB signaling pathway’s expression, further enhancing the number of inflammatory cytokines in the LPS-induced MLE-12 cells, thus aggravating the acute lung injury. We further showed that salidroside could effectively reverse this effect and alleviate endotoxin-induced acute lung injury.
Multiple studies have shown that miRNA was related to the onset and development of lung cancer, pulmonary tuberculosis, ALI, pulmonary fibrosis, and other lung diseases.28–30 During inflammatory and oxidative stress responses, different miRNAs promote or inhibit inflammation and oxidative stress. 31 MiR-124-5p overexpression can inhibit NOX-mediated ROS production and NF-κB signaling pathway activity, thus, improving the cerebral ischemia-reperfusion injury. 32 When MiR-885-3p was overexpressed in the peripheral blood mononuclear cells in Type 1 diabetes patients, it inhibited their inflammatory response as it targeted the regulation of the TLR4/NF-κB signaling pathway. 33 Studies have also shown that the exosome miR-199a-5p from the endothelial cells can reduce apoptotic activity and the inflammatory responses in the nerve cells by regulating the stress in the endoplasmic reticulum (ER). 34 MiR-199a-5p can regulate excessive lung inflammation in cystic fibrosis by regulating the CAV1 pathway. 35 MiR-199a-5p regulates the TNF-α and IL-6 generation in the human synovial fibroblasts by regulating the P38, ERK, and JNK signaling pathways. 36 However, none of these studies have described the mechanism used by miRNA-199a-5p for regulating the inflammatory response in the cells. Here, we demonstrated that the miR-199a-5P overexpression decreased TNFAIP8L2 expression and increased the expression of the HMGB1/TLR4/NF-κB signaling pathway, thereby increasing the inflammatory response of the MLE-12 cells, which further aggravated the acute lung injury. A lower miR-199a-5p expression enhanced the TNFAIP8L2 expression, reduced the HMGB1/TLR4/NF-κB signaling pathway activity, and inhibited the inflammatory response of MLE-12 cells, finally assuaging the acute lung injury that was induced by sepsis.
To clarify the mutual regulatory relationship between miR-199a-5p and TNFAIP8L2, we constructed wild-type and mutant TNFAIP8L2 and performed a dual luciferase reporter gene assay to detect whether TNFAIP8L2 is a regulatory target of miR-199a-5p. We found that TNFAIP8L2 was the regulatory target of miR-199a-5p, which suggests that miR-199a-5p regulates the HMGB1/TLR4/NF-κB inflammatory pathway by targeting the expression of TNFAIP8L2 and hence regulating LPS-induced MLE-12 cells inflammation.
Studies show that salidroside prevents TNF-α–induced cardiac microvascular endothelial cell inflammation, as it blocks the activity of the mitogen-activated protein kinase and activates the NF-κB signaling pathway. 37 Salidroside improves the endothelial inflammatory response and reduces oxidative stress by controlling the AMPK/NF-κB/NLRP3 signaling pathway in the human umbilical vein endothelial cells induced by the glycation end-products. 38 Salidroside also alleviates the furan-induced liver inflammation in mice by moderating the oxidative and ER stress. 39 Furthermore, salidroside has been shown to alleviate airway inflammation and remodeling in asthmatic mice by suppressing the expression of the miR-323-3p in the cytokine signaling 5 (SOCS5). 40 In this study, we showed that salidroside inhibited miRNA-199a-5p, increased the TNFAIP8L2, decreased the HMGB1/TLR4/NF-κB signaling pathway, decreased the secretion of inflammatory cytokine molecules in the MLE-12 cells, and assuaged the endotoxin-induced acute lung injuries.
Limitations
This study examined the protective mechanism of salidroside modulating miR-199a-5p/TNFAIP8L2 on lipopolysaccharide-induced MLE-12 cells. However, several limitations exist in this study. Firstly, the effect of miR-199a-5p on TNFAIP8L2 expression in MLE-12 cells may not be limited to inflammation and apoptosis-related protein pathways. Oxidative stress, ferroptosis, autophagy, pyroptosis, and other proteins and pathways might also be involved. Secondly, in addition to the TNFAIP8L2/HMGB1/TLR4/NF-κB pathway, other pathways might also be associated with the protective effects of salidroside against LPS-induced MLE-12 cells. The protective effect of salidroside might involve oxidative stress, apoptosis, pyroptosis, and other processes. Further investigation must be conducted with in vivo animal models to determine the underlying mechanisms. Moreover, this study also did not predict other target genes of the miRNA.
Conclusions
In this study, we showed that miR199-a-5p targeted the regulation of TNFAIP8L2. It also regulated the endotoxin-induced acute lung injury by targeting the activity of the HMGB1/TLR4/NF-κB signaling pathway. Salidroside decreased the miR-199a-5p, increased the TNFAIP8L2, curbed the HMGB1/TLR4/NF-κB signaling pathway, decreased inflammatory cytokines secretion in the MLE-12 cells, and assuaged the endotoxin-induced acute lung injury. We also observed the protective mechanism of salidroside against sepsis-induced acute lung injury and presented a novel theoretical basis and a different therapeutic agent for salidroside while treating sepsis-induced acute lung injuries.
Footnotes
Acknowledgments
The authors are grateful to Enpaper for proofreading the English text of an earlier version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Yunnan Provincial Department of Education Project (No. 2019J1228) for funding this study.
Trial registration
Trial registration number: KY20180714.