
Abstract
Objectives
Platelet-rich plasma (PRP) plays an important role at all stages of wound healing, including the inflammatory stage. Macrophage autophagy has been found to influence the inflammatory response process. However, it is unclear whether PRP can affect inflammatory responses via macrophage autophagy. In the present study, we explored the effect of PRP on inflammatory responses and researched the underlying mechanism.
Methods
RAW 264.7 macrophages were treated with PRP and/or lipopolysaccharide (LPS). The effects of PRP on the expression of inflammatory factors were determined by ELISA and qRT-PCR. Macrophage autophagosomes were also assessed by TEM and immunofluorescence. Autophagy and NLRP3-related proteins were investigated using Western blot analysis.
Results
PRP reduced the levels of inflammatory factors and increased autophagy in RAW 264.7 cells. Pretreatment with 3-MA, which is an autophagy inhibitor, abolished the impact of PRP on the inflammatory response. Moreover, PRP induced macrophage autophagy by activating the NLRP3 inflammasome.
Conclusions
These results show that PRP can attenuate LPS-induced inflammatory responses by enhancing autophagy via NLRP3. These study also provides a new perspective on the molecular mechanism of PRP therapy in wound healing.
Introduction
Platelet-rich plasma (PRP) is a platelet concentrate extracted from whole blood by centrifugation. Activated PRP releases growth factors, cytokines and antibacterial peptides, which stimulates regenerative processes including hemostasis, inflammation, regeneration and immune function. 1 PPR has been found to modulate the function of a wide variety of cells involved in tissue repair. Moreover, PRP regulates a variety of cellular functions including macrophages in tissue repair.2,3
The inflammatory response is a kind of defensive physiological response that involves cytokines, chemokines and lipid inflammatory mediators. Macrophages are considered to be one of the most important immune cells in the whole process of inflammation. 4 Macrophages play a key role in inflammatory responses by quickly reaching injury sites, killing pathogens and secreting cytokines. 5 Activating macrophages by lipopolysaccharide (LPS) can produce proinflammatory factors and strongly induce the inflammatory reaction. 6 Therefore, macrophages have been proposed to be potential targets for regulating the body’s inflammatory responses. It has also been found that platelets can regulate macrophage polarization. Furthermore, platelet microparticles have been shown to transform monocytes into M2 macrophages, which can reduce inflammation and perform tissue repair.7,8 PRP can reduce inflammatory factor levels and increase the expression of anti-inflammatory factors in mice.9,10 Based on these findings, we hypothesized that PRP could improve LPS-induced inflammatory responses in macrophages. In support of this, a recent study demonstrated that platelet-rich fibrin (PRF) had anti-inflammatory activity. 11 PRF is thought to be a downstream product of PRP, which is used in the natural coagulation mechanism to form fibrin clots. 12 However, unlike PRP, PRF cannot regulate platelet concentrations and has been mainly applied in regenerative dentistry. 13 Moreover, PRF has low extraction efficiency, meaning little can be produced at one time. 13 Thus, we choose PRP instead of PRF to study its role in inflammatory responses.
Autophagy is a ubiquitous cellular process that maintains intracellular homeostasis by using lysosomes to degrade damaged organelles and macromolecules. 14 Autophagy helps suppress inflammasomes and excessive inflammatory activation,15,16 and attenuates the LPS-induced secretion of inflammatory factors. 17 In addition, PRP can protect chondrocyte function by increasing chondrocyte autophagy and the secretion of anti-inflammatory factors. 18 Therefore, identifying the factor in PRP that plays a role in directing inflammatory responses through autophagy ought to be investigated.
Although numerous studies have shown that PRP plays a role in the development of inflammation, its mechanism remains unclear. The purpose of this study was to explore the impacts of PRP on the inflammatory responses induced by LPS and to investigate its underlying mechanism.
Materials and methods
Materials
Anti-GAPDH and anti-LC3 antibodies were purchased from Cell Signaling Technology Inc (Massachusetts, USA). Anti-Bclin1 and anti-P62 antibodies were purchased from Santa Cruz Biotechnology (California, USA). Anti-NLRP3 antibodies were obtained from R&D Systems (Minnesota, USA). Anti-Pro-IL-1β, anti-IL-1β, anti-Pro-caspase-1, and anti-caspase-1 antibodies were purchased from Abcam Technology (Massachusetts, USA). TNF-α, TGF-β, IL-10, and IL-1β-ELISA kits were obtained from RayBiotech (Georgia, USA). 3-MA, LPS, penicillin and streptomycin were obtained from Sigma-Aldrich (Missouri, USA). Horseradish peroxidase-conjugated second antibodies, bicinchoninic acid protein assay kits and nitric oxide assay kits were obtained from the Beyotime Institute of Biotechnology (Beijing, China). Total RNA Extraction kits were purchased from Promega Corporation (Wisconsin, USA). High-capacity cDNA reverse transcription kits and Maxima TMSYBR Green/ROX qPCR Master Mix (2X) were obtained from Thermo Fisher Scientific (Massachusetts, USA)
Methods
The nature of our study is: cell model in vitro.
Preparation of platelet-rich plasma
Blood samples from a total of 12 type O healthy voluntary unpaid blood donors were collected from the Blood Center of the Southern Theater Command from January to October 2020. Written informed consent was obtained from blood providers. (Inclusion criteria: Male, aged 25–35 years old, Hemoglobin ≥120 g/L, Platelet count >120×109/L. Exclusion criteria: Have an infectious disease (HIV, TP, HBV, HCV), blood-related diseases or severe cardiovascular disease. Taking aspirin or aspirin-like drugs within 3 days.) All samples were tested for blood-related infectious diseases according to Chinese testing standards. 19 Samples were then centrifuged at 510 g for 20 min, and the supernatant was saved as platelet-poor plasma (PPP). Most of the PPP was then removed, and PRP was resuspended in the remaining PPP to reach a concentration of approximately 1.0 × 1012 platelets/L. The PRP was next activated by thrombin, and the supernatant was collected after centrifugation for 10 min at 2826 g. The resulting PRP supernatant was cryopreserved at −80°C until further use.
Cell culture
RAW264.7 cells were obtained from the Established of Organic chemistry and Cell Natural chemistry and Cell Science (SCSP-5036, Shanghai, China). Cells were maintained in DMEM supplemented with 10% fetal bovine serum at 37°C in a humidified chamber in 5% CO2. For different experiments, RAW264.7 cells were treated with LPS (100 ng/mL) for 24 h, followed by treatment with PRP (10%), PPP (10%) or vehicle for an additional 24 h. To inhibit autophagy, 3-MA (5 mM) was added for 1 h.
ELISA
RAW264.7 cells were first seeded in 96 well plates at 1.5 × 104/well. Cultured cell supernatant was collected after the treatments mentioned in Cell culture and then centrifuged for 10 min at 177 g. The cell supernatant was then transferred to a fresh tube and stored at −80°C. Before experiments, the supernatant was incubated in a 37°C water bath. Next, 100 μL of standard or sample was added to ELISA wells at room temperature (RT) for 2.5 h, followed by the addition of a prepared biotin antibody at RT for 1 h. After adding 100 μL of prepared streptavidin solution for 45 min, TMB One-Step substrate reagent was added and plates were incubated for 30 min at RT. Following three washes, 50 μL of stop solution was added, and the optical density of each well was read at 450 nm immediately. For each experiment, a standard curve was generated by plotting mean absorbance against protein concentration. The concentration of each sample was calculated by using this standard curve.
Nitric oxide determination
Nitric oxide (NO) secreted by RAW264.7 macrophages was quantified using the Griess reaction. Cells and supernatant were prepared the same as described previously. Standard or sample was added to a 96 well plate at 50 μL/well from low to high, followed by Griess I and II, respectively. The absorbance was then measured at 540 nm, a standard curve was drawn based on the absorbance value of a standard product, and the sample concentration for each sample was calculated using this curve.
Western blot analysis
RAW264.7 macrophages were seeded in six well plates at 5 × 105/well. After all treatments, cells were lysed with RIPA buffer and PMSF. The protein concentration was determined using a BCA protein assay kit. Proteins were then separated using 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were next incubated with anti-P62 (1:1000), anti-Beclin 1 (1:1000), anti-LC3 (1:1000), anti-NLRP3 (1:1000), anti-Pro-IL-1β (1:1000), anti-IL-1β (1:1000), anti-Pro-caspase-1 (1:1000), anti-caspase-1 (1:1000), or GAPDH antibodies (1:2000) overnight at 4°C, washed and then incubated with secondary antibody (1:5000) for 1 h at RT. An enhanced chemiluminescence detection kit was used to visualize the protein bands. The Scion Image software was used to perform Densitometric analysis.
RNA preparation and real-time PCR (qRT-PCR)
Primer sequences for real-time PCR.

Immunofluorescence
RAW264.7 cells were first fixed and treated with absolute ethyl alcohol for 30 min. Goat serum was used for blocking at RT for 1 h. Slides were next incubated with anti-LC3 (1: 200) at 4°C overnight. After recovery at room temperature, a fluorescent secondary antibody (Alexa Fluor 488-conjugated goat anti-rabbit IgG, 1:1000) was added for 1 h. DAPI (1:1000) was used to counterstain the nucleus for 1 min. Cells were finally imaged by fluorescence microscopy.
Transmission electron microscopy
Three% glutaraldehyde was first used to fix RAW264.7 cells at RT for 8 hours. After fixation, cells were treated with 1% osmium tetroxide and dehydrated from 50% to 100% ethyl alcohol, and cells were embedded with Epon-812 and sliced using an ultramicrotome. Three percent uranium acetate and lead citrate were next used to double-stain sections. Morphology was then observed, and photos were taken by transmission electron microscopy.
Statistical analysis
Statistical analysis was performed using the Mann–Whitney U test and Kruskal–Wallis test, followed by Dunn’s multiple comparison test. All experiments were performed independently three to six times. Analysis was performed using SPSS.22.0. p ≤ 0.05 was considered statistically significant.
Results
Platelet-rich plasma improves lipopolysaccharide-induced inflammatory responses in RAW264.7 cells
To verify the effect of PRP on macrophage inflammatory responses, RAW264.7 cells were treated with PRP, PPP and/or LPS for 24 h. For this, the levels of TNF-α, IL-1β, NO, IL-10 and TGF-β were measured by the Griess reaction and ELISA. PRP significantly increased the levels of IL-10 and TGF-β compared with the control group (p < 0.05) and partially abrogated the inhibitory effect of LPS on the protein expression of these two markers. Furthermore, PRP decreased the levels of IL-1β, TNF-α and NO that were induced by LPS (p < 0.05). PPP, by contrast, did not significantly affect the expression of these markers (p > 0.05) (Figure 1(a)). The gene expression of TNF-α and IL-1β was next quantified by qRT-PCR. LPS significantly increased the expression of these markers, whereas PRP attenuated this stimulatory effect. Moreover, PRP significantly increased Arg-1 and IL-10 expression (Figure 1(b)). These results indicated that PRP had an exceptional impact on decreasing inflammatory responses. PRP improves LPS-induced inflammatory responses in RAW264.7 cells. RAW264.7 cells were treated with LPS (100 ng/mL) for 24 h, followed by treatment with PRP (10%), PPP (10%) or vehicle for an additional 24 h. (a) The expression of TNF-α, TGF-β, IL-10, IL-1β and NO was assayed by ELISA. (b) mRNA levels of TNF-α, IL-1β (M1-related markers), IL-10 and Arg-1 (M2-related markers) were detected by qRT-PCR. Values are presented as the mean ± SD. N = 3, *p < 0.05 versus the control group; #p < 0.05 versus the LPS group.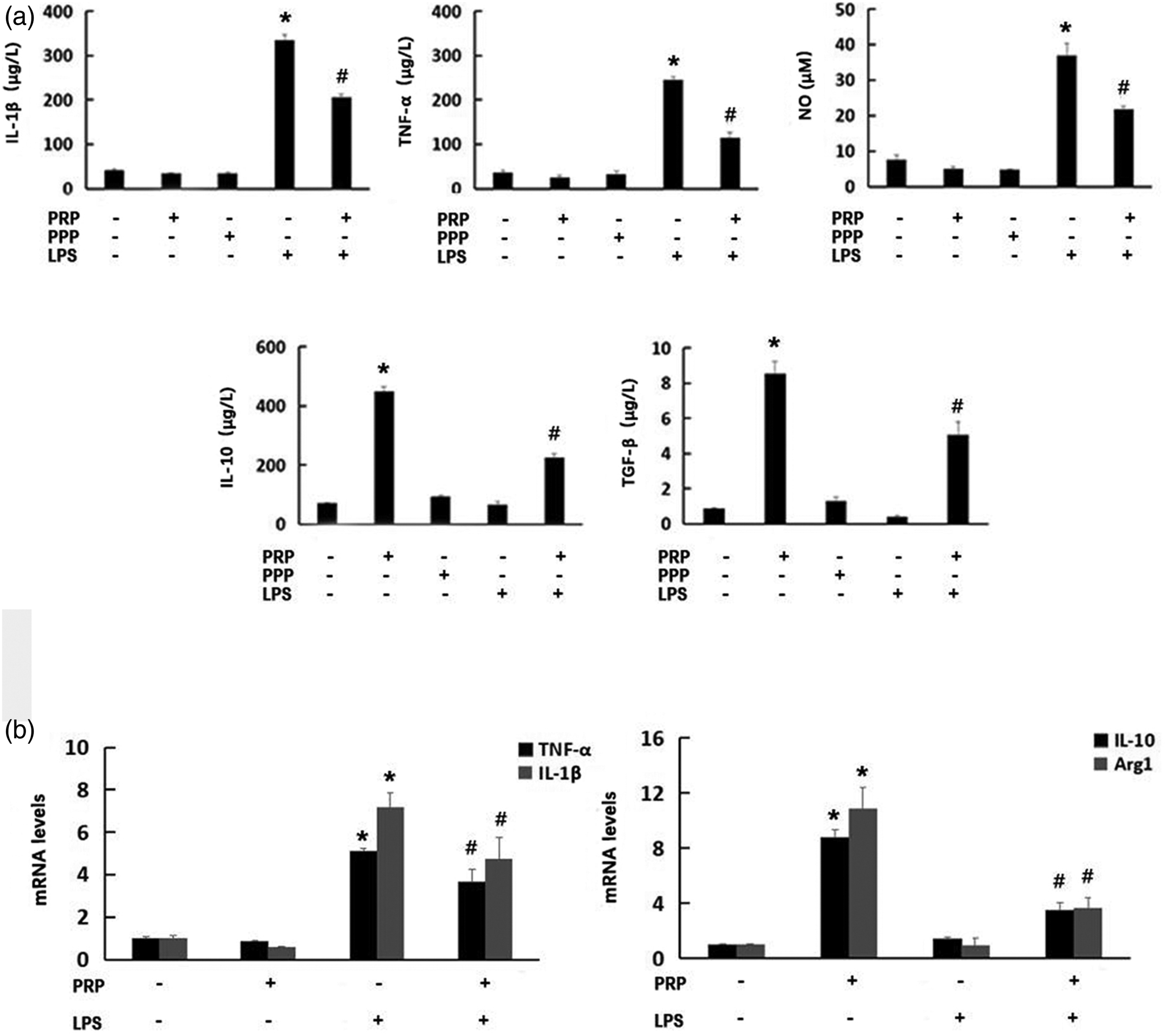
Platelet-rich plasma induces macrophage autophagy in RAW264.7 cells
p62, Beclin-1 and LC3 levels were next determined by Western blotting to investigate whether PRP modulates autophagy in macrophages. PRP decreased the p62 abundance and increased the ratio of LC3II to LC3I and Beclin-1 (Figure 2(a) and (b)). Macrophages were treated with 3-MA for 2 hours in the presence of PRP, and 3-MA inhibited PRP-induced autophagy. Autophagosome formation was also assessed by TEM, which is the most commonly used method for qualitative and quantitative analysis of autophagy, and the results showed that PRP induced the formation of vacuoles in macrophages. Autophagic cells were also identified by immunofluorescence with the autophagy marker LC3, and the results revealed that PRP significantly increased LC3 expression (Figure 2(b) and (c)). These findings suggest that PRP can increase autophagy in RAW264.7 cells. PRP-induced macrophage autophagy in RAS264.7 cells. (a) RAW264.7 cells were pretreated with 3-MA (5 mM) for 1 h, followed by treatment with or without PRP (10%). Total RAW264.7 cell lysates were immunoblotted with anti-p62, anti-Beclin1, anti-LC3 and anti-GADPH antibodies. (b) Representative TEM images depicting the ultrastructure of RAW264.7 cells incubated with PRP (10%) or vehicle for 24 h. Autophagosomes are indicated with arrows. (c) Cells were treated with PRP (10%) or vehicle for 24 h. Cells were moreover stained for LC3 (green) and with DAPI (blue), and visualized by fluorescence microscopy (400×). Values are presented as the mean ± SD. N = 3, *p < 0.05 versus the control group; #p < 0.05 versus the 3-MA group.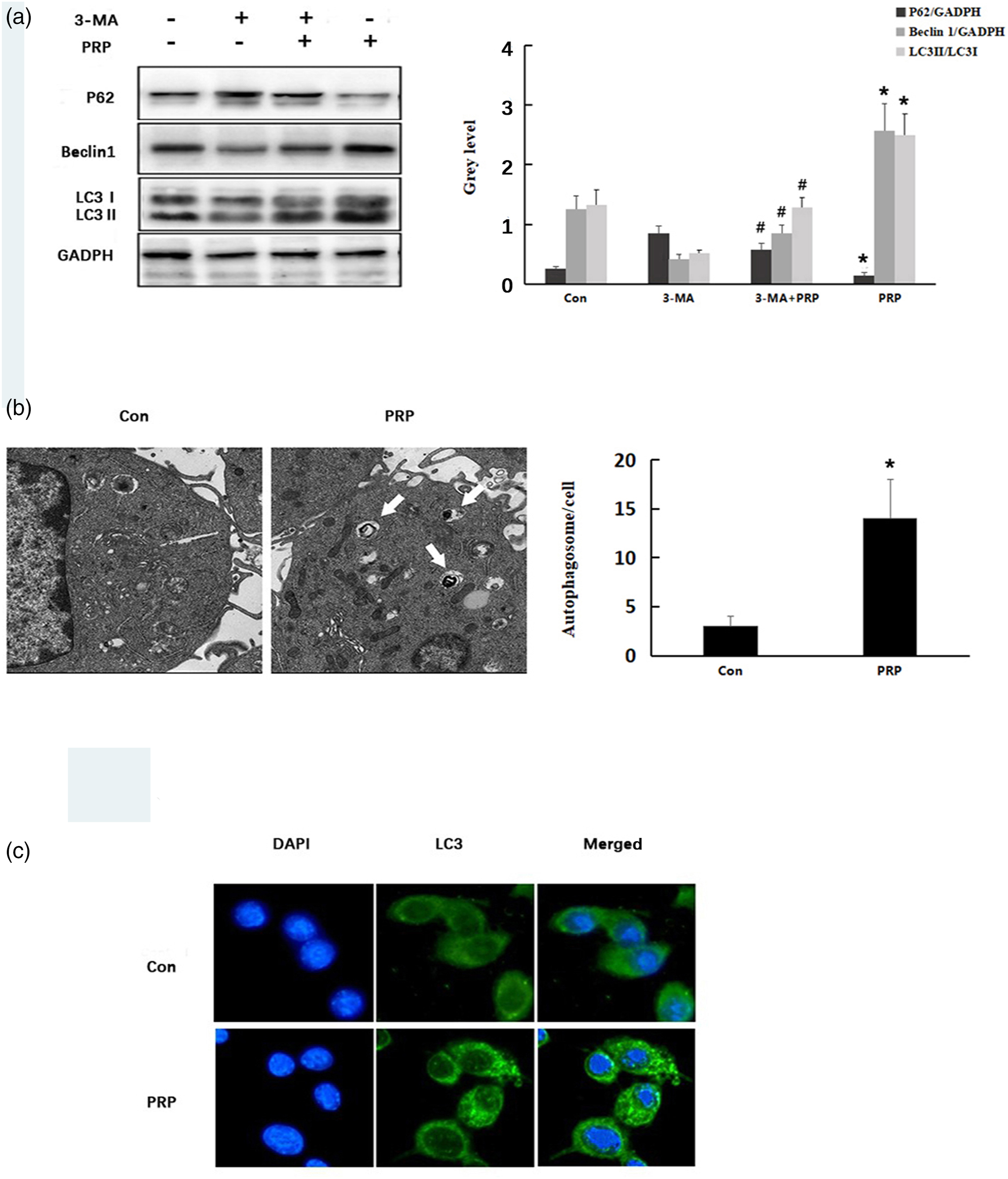
Platelet-rich plasma improves lipopolysaccharide-induced inflammatory responses by inducing autophagy in RAW264.7 cells
To determine whether autophagy affects PRP improves LPS-induced inflammatory response by inducing autophagy in RAW264.7 cells. RAW264.7 cells were pretreated with 3-MA (5 mM) for 1 h and then treated with or without LPS (100 ng/mL) for 24 h, followed by stimulation with PRP (10%) for an additional 24 h. (a) The expression of TNF-α, TGF-β, IL-10, IL-1β and NO was assayed by ELISA. (b) mRNA levels of TNF-α, IL-1β (M1-related markers), IL-10 and Arg-1 (M2-related markers) were detected by qRT-PCR. Values are presented as the mean ± SD. N = 3, *p < 0.05 versus the PRP group; #p < 0.05 versus the LPS and PRP co-treated group.
Platelet-rich plasma induces autophagy through the NLRP3 inflammasome
To investigate whether PRP induces autophagy through the NLRP3 inflammasome, PRP and 3-MA were used to treat LPS-simulated RAW264.7 cells. The levels of NLRP3, IL-1β, and caspase-1 were thus assessed by Western blot (Figure 4). The analysis indicated that NLRP3, IL-1β, and caspase-1 levels were markedly decreased in the PRP therapy group compared with those in the LPS group, whereas 3-MA disturbed the effect of PRP (p < 0.05). These data suggested that NLRP3 activation played a key role in PRP-induced autophagy. PRP induces autophagy through the NLRP3 inflammasome in RAW264.7 cells. RAW264.7 cells were pretreated with 3-MA (5 mM) for 1 hour and then treated with or without LPS (100 ng/mL) for 24 h, followed by stimulation with PRP (10%) for an additional 24 h. (a) Representative images and quantitation of Western blots showing the protein expression of NLRP3, Pro-IL-1β, IL-1β, Pro-caspase-1 and caspase-1. (b) Representative images and quantitation of Western blots showing the protein expression of Pro-IL-1β, IL-1β, Pro-caspase-1 and caspase-1. Values are presented as the mean ± SD. N = 3, *p < 0.05 versus the control group; **p < 0.05 versus the LPS treated group; ##p < 0.05 versus the LPS and PRP co-treated group.
Discussion
In recent years, PRP, as an effective treatment means, has become widely used in regenerative medicine. 20 Studies have shown that PRP plays a role in all stages of wound healing, including the inflammatory phase. 21 In the present study, PRP improves LPS-induced inflammatory responses by inducing autophagy, and the NLRP3 inflammasome may play a role in this. The inflammatory response involves the soluble proinflammatory cytokines and the inflammatory mediators produced by immune cells, and is used in combating infections and damage caused by external factors. 22 Macrophages participate in all phases of the inflammatory response and, upon tissue damage, are recruited to the site of injury. Macrophages phagocytose pathogenic microorganisms, remove necrotic fragments at the sites of injury, and secrete bioactive substances such as IL-1β, TNF-α, and NO. However, excessive proinflammatory factor secretion can lead to dysregulated inflammatory responses, infection and limited tissue repair. As is well known, LPS regulates the inflammatory signaling cascade and the emission of proinflammatory cytokines. 23 Previous studies have confirmed that PRP can significantly reduce the gene expression and secretion of IL-6, 24 and platelets reduce the LPS-induced secretion of TNF-α, IL-6, and NO by macrophages. PRP has also been found to significantly downregulate the level of LPS-induced proinflammatory genes. 25 In our study, PRP induced the expression of IL-10 and TGF-β, but it did not significantly affect the expression of IL-1β, TNF-α, or NO. Furthermore, PPP had no significant effect on the release of inflammatory factors. These results suggest that PRP can inhibit excessive inflammation. The most likely is due to various biomolecules from activated platelets.
Autophagy is a complex process that degrades dysfunctional cellular components within cells via lysosomes. 26 Several proteins participate in autophagy, protecting cells by engulfing and killing pathogens, and decreasing the production of proinflammatory cytokines. 27 Thus, autophagy can inhibit the inflammatory reaction and reduce the inflammatory tissue damage produced by disease. Our results showed that PRP increased the expression of LC3II and Beclin-1 in macrophages and reduced the expression of the autophagic substrate p62. Electron microscopy and immunofluorescence staining showed that PRP increased autophagosome numbers, suggesting that PRP promoted autophagy in macrophages. Autophagy can promote neovascularization during wound healing by regulating the secretion of VEGF, 28 and PDGF increases the expression of autophagy-related genes, 29 demonstrating that autophagy is closely regulated by the growth factors secreted by platelets, which may explain why PRP promotes autophagy.
Autophagy plays an important regulatory role in macrophage inflammatory responses. A previous study showed that adiponectin could induce autophagy by inhibiting the expression of the proinflammatory factors IL-1β and TNF-α in macrophages. 30 Here, we showed that PRP-induced autophagy stimulated M2-related gene expression and CD206-positive cell levels. In contrast, autophagy inhibition reduced PRP-induced M2 polarization. These findings demonstrate that autophagy plays an important role in PRP-induced regulation of inflammatory responses.
NLRP3 is a typical representative of the NALPs protein family and is mainly expressed in neutrophils and macrophages. 31 The inflammasome can regulate caspase-1 activation and activated caspase-1 further promotes the maturation and secretion of the precursor pro-IL-1β, inhibits phagocytosis, induces inflammatory innate immune responses and plays a key role in host defense against infectious substances. 32 Excessive secretion of inflammatory cytokines can lead to apoptosis, thereby inducing the occurrence of related diseases.33,34 NLRP3 inflammasome expression in macrophages was also shown to be up-regulated by LPS via autophagy activation. 35 In addition, our results showed that NLRP3, IL-1β and caspase-1 levels were markedly decreased in the PRP therapy group compared with those in the LPS group, whereas 3-MA disturbed the effect of PRP. These results suggest that NLRP3 activation plays a key role in PRP-induced autophagy modulation.
Conclusions
In conclusion, we suggest that autophagy plays an important role in the anti-inflammatory effects of PRP. Moreover, the NLRP3 inflammasome may be related with PRP-induced anti-inflammatory responses. Our study demonstrated that PRP improves LPS-induced inflammatory responses by decreasing NLRP3 inflammasome levels by upgrading autophagy (Figure 5). These findings provide new insights into the PRP regulation of inflammation and lay a foundation for further mechanistic research in the future. The limitations of the study were that we did not calculate and justify the sample selected and just use three samples in each group for the experiments. Besides, we only conducted in vitro, which may be different from the inflammatory responses in vivo. In future experiments, the effect of PRP on macrophage polarization and its mechanism in the inflammatory response in vivo will be further investigated. PRP improves LPS-induced inflammatory responses by autophagy. After exposing cells to LPS, the inflammation of macrophages was increased. However, our study showed that PRP induces macrophage autophagy to suppress LPS-induced inflammation. Moreover, PRP regulates macrophage autophagy to inhibit the NLRP3 inflammasome, which is induced by LPS and leads to inflammation. (The figure was drawn by the author).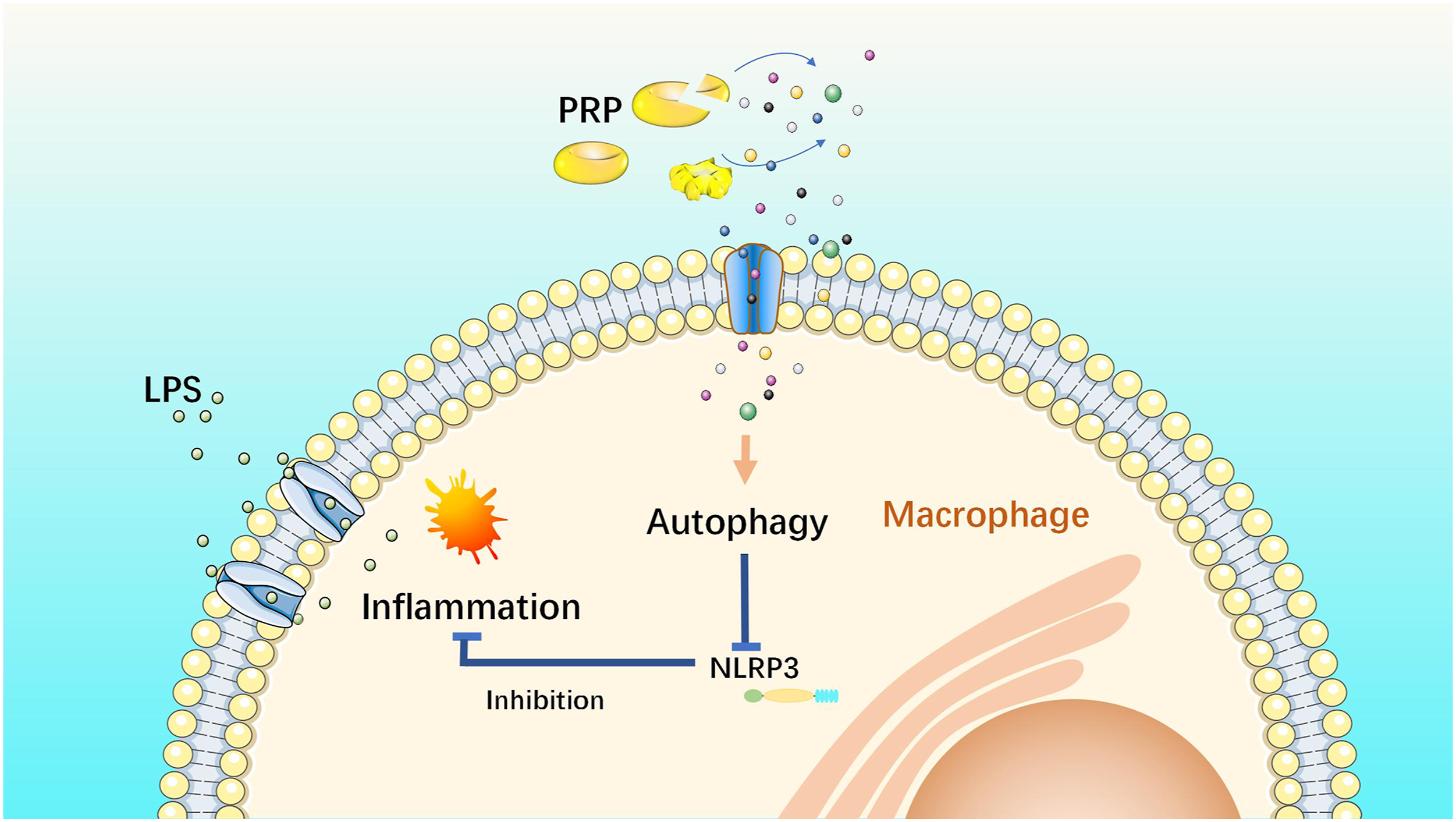
Footnotes
Acknowledgements
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grants from the National Natural Science Foundation of China (NSFC, No. 81701913).
Ethics approval
Ethical approval for this study was obtained from the Research Ethics Committee of the General Hospital of the Southern Theatre Command (NZLLKZ2022064)
Informed consent
Written informed consent was obtained from blood providers before the study.