
Abstract
Coronary artery disease has remained a major health challenge despite enormous progress in prevention, diagnosis, and treatment strategies. Formation of atherosclerotic plaque is a chronic process that is developmentally influenced by intrinsic and extrinsic determinants. Inflammation triggers atherosclerosis, and the fundamental element of inflammation is the immune system. The immune system involves in the atherosclerosis process by a variety of immune cells and a cocktail of mediators. It is believed that almost all main components of this system possess a profound contribution to the atherosclerosis. However, they play contradictory roles, either protective or progressive, in different stages of atherosclerosis progression. It is evident that monocytes are the first immune cells appeared in the atherosclerotic lesion. With the plaque growth, other types of the immune cells such as mast cells, and T lymphocytes are gradually involved. Each cell releases several cytokines which cause the recruitment of other immune cells to the lesion site. This is followed by affecting the expression of other cytokines as well as altering certain signaling pathways. All in all, a mix of intertwined interactions determine the final outcome in terms of mild or severe manifestations, either clinical or subclinical. Therefore, it is of utmost importance to precisely understand the kind and degree of contribution which is made by each immune component in order to stop the growing burden of cardiovascular morbidity and mortality. In this review, we present a comprehensive appraisal on the role of immune cells in the atherosclerosis initiation and development.
Introduction
The growing trend of cardiovascular morbidity and mortality imposes a huge financial and social toll. 1 Most of the cardiovascular-related devastating sequels are attributed to coronary artery disease (CAD). The main cause of CAD is atherosclerosis, which is developed by a complex interplay between intrinsic and extrinsic factors. 2 Immune system is among the intrinsic factors that contributes substantially to the formation and development of atherosclerosis inasmuch as some researchers consider atherosclerosis as an autoimmune disease. 3 Atherosclerosis development is mainly determined by the balance between inflammatory and anti-inflammatory features. Meanwhile, other CAD risk factors like hyperlipidemia is potentially able to recruit immune cells, and orient the atherosclerosis progress in favor of enhanced plaque formation. 2 Indeed, individuals with metabolic syndrome and obesity are exposed to a variety of cytokines that exacerbate the condition.4,5 Adipose tissue itself releases some cytokines including leptin, adiponectin, and resistin which augment the inflammatory reactions. 4 Peroxidation of the lipids within adipose tissue generates molecules with both inflammatory and anti-inflammatory functions which act through binding to nuclear receptors controlling inflammation-related genes.6,7
Atherosclerosis is probably initiated by entrapment of low-density lipoprotein (LDL) in the intimal layer of the coronary arteries. In the interim, endothelial injury provides suitable circumstances for circulating monocytes to gain access to the subendothelial layer. Some impairments like hyperlipidemia, hypertension, blood flow disorders, and increased level of oxidative stress facilitate the occurrence of endothelial injury. Monocytes transform to macrophages after receiving some stimuli from surroundings. The newly formed macrophages engulf modified LDLs generating cells with lipid-laden foamy appearance. After several cycles of engulfing, macrophages die due to extensive uptake of lipid particles. Accumulation of dead cells establishes a necrotic core. Finally, smooth muscle cells (SMCs) cover this core, and the atherosclerotic plaque with a fibrous cap is formed
8
(Figure 1). Notably, the aforementioned order of events are according to what happens in the humans, and there may be some differences in other species. Schematic presentation of atherosclerotic plaque formation with the involvement of major players. TNF-α, Tumor necrosis factor alpha; IL-6, Interleukin 6; IL-8, Interleukin 8; IFN-γ, Interferon gamma; MCSF, Macrophage colony-stimulating factor; LDL, Low density lipoproteins; Ox-LDL, Oxidized low-density lipoprotein; HDL, High density lipoproteins; TLR-2, Toll-like receptor 2; TLR-4. Toll-like receptor 4; CD14, Cluster of differentiation 14.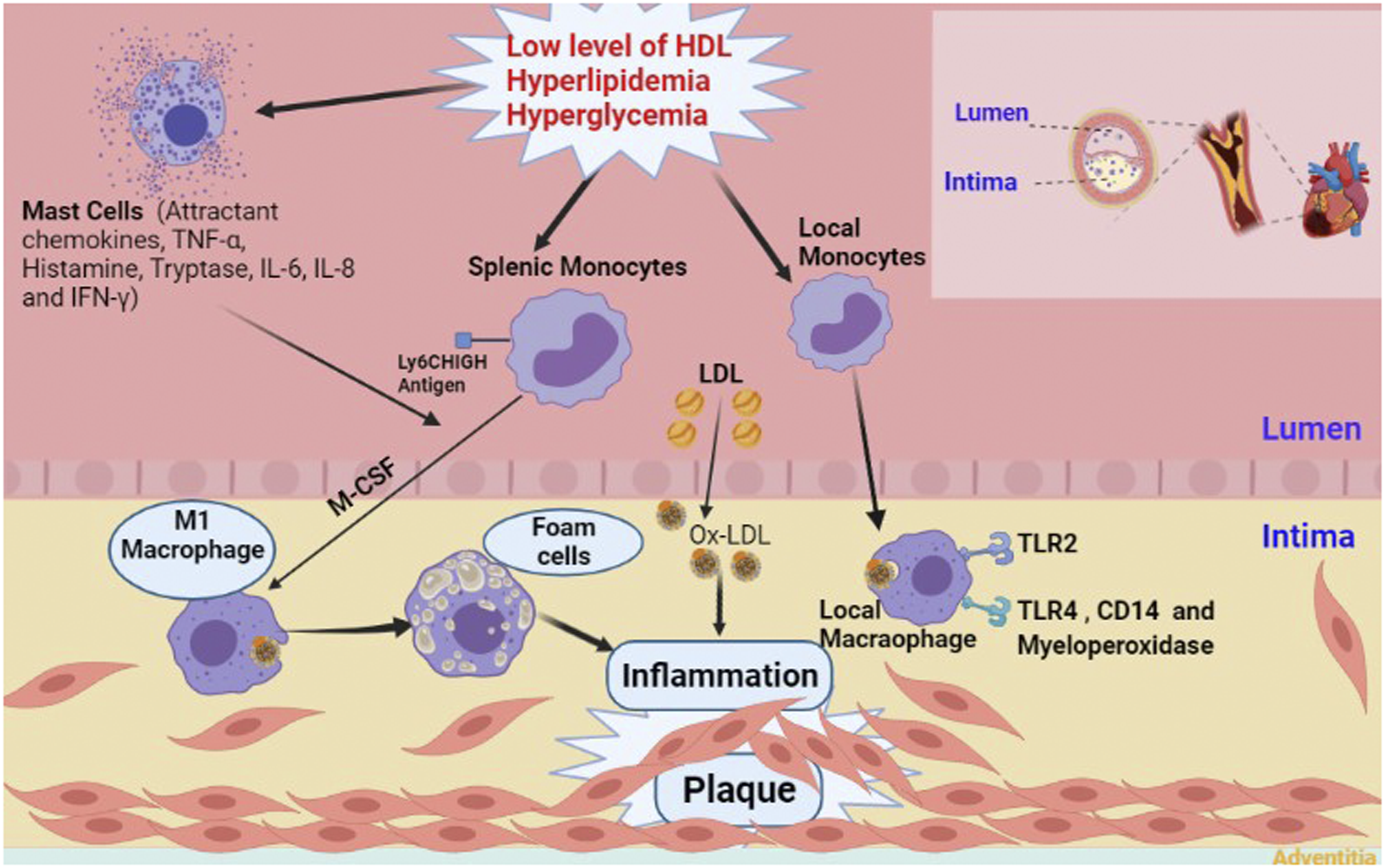
In this review, we tried to present a comprehensive appraisal on the role of immune system in the atherosclerosis process in order to seek how immune cells interact with every component of the plaque as well as with each other toward formation and development of CAD.
Contribution of the immune system to atherosclerosis: From the first step to the last
Innate and adaptive immunity play a significant role in the atherosclerosis process. 9 Immune cells harbor pattern recognition receptors (PRRs) on their surface, and scrutinize different organs for detecting pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) in order to eliminate pathogens or damaged cells, respectively.10,11 In the early stages of atherosclerosis, modified endogenous molecules provide antigens for antigen presenting cells promoting activation of adaptive immunity. 12 LDL particles, which are modified in different ways, play the role of immune complexes,13,14 and make up immunogenic entities. For example, epitopes of oxidized LDL (oxLDL) are one of the major DAMPs in the subendothelial space that are recognized by PRRs of the immune cells like toll-like receptors (TLR). 15 Indeed, it is believed that LDL stimulates the expression of these receptors corroborating high amounts of TLR-4 in human atherosclerotic plaques.16,17
Also, macrophages use their receptors, surface CD1 molecule and major histocompatibility-complex (MHC) molecules, to present ingested LDLs as lipid and peptide antigens, respectively. This presentation activates a member of adaptive immune system, CD4+ T cells. 18 Encountering of naïve T cells with antigens transform them into effector (memory) T cells, in particular Th1 cells producing IFN-γ, a pro-atherogenic cytokine. 18 In addition to macrophages, IFN-γ, TNF-α, and IL-6 are secreted by other immune cells like mast cells and neutrophils. 19 These inflammatory mediators with possible proangiogenic characteristics 20 upregulate the expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) on endothelial cells. 19 Increasing local concentration of inflammatory mediators recruit further immune cells to the region, and totally, lead to the set-up of a chronic inflammation with accumulation of dead cells. 18 Immune cells also release matrix metalloproteinases that degrade extracellular matrix (ECM), and then promote reconstruction and neoangiogenesis of the ECM which results in the shift of protein content in the ECM during atherosclerosis progression.21–23
However, expression of strong anti-inflammatory cytokines besides appearance of antibody responses by B cells have significant impacts on the counterbalancing of the atherosclerotic-related immune reactions.
2
For instance, interleukin 10 and TGF-β tightly control T cell responses as the absence of these two cytokines augments the progress of atherosclerosis.
2
Due to the profound involvement of the immune system in atherosclerosis, some studies proposed targeting immune components as a strategy to slow down the rate of plaque formation. Canakinumab, a monoclonal antibody against IL-1β, decreased the rate of recurrent cardiovascular events by targeting overactive immune system without lowering serum lipid level.
24
However, different components of the immune system play contradictory roles in relation to atherosclerosis progression (Figures 2 and 3). Moreover, restrain of the immune system substantially decreases natural defense mechanisms against pathogenic invaders increasing the risk of fatal infections.
25
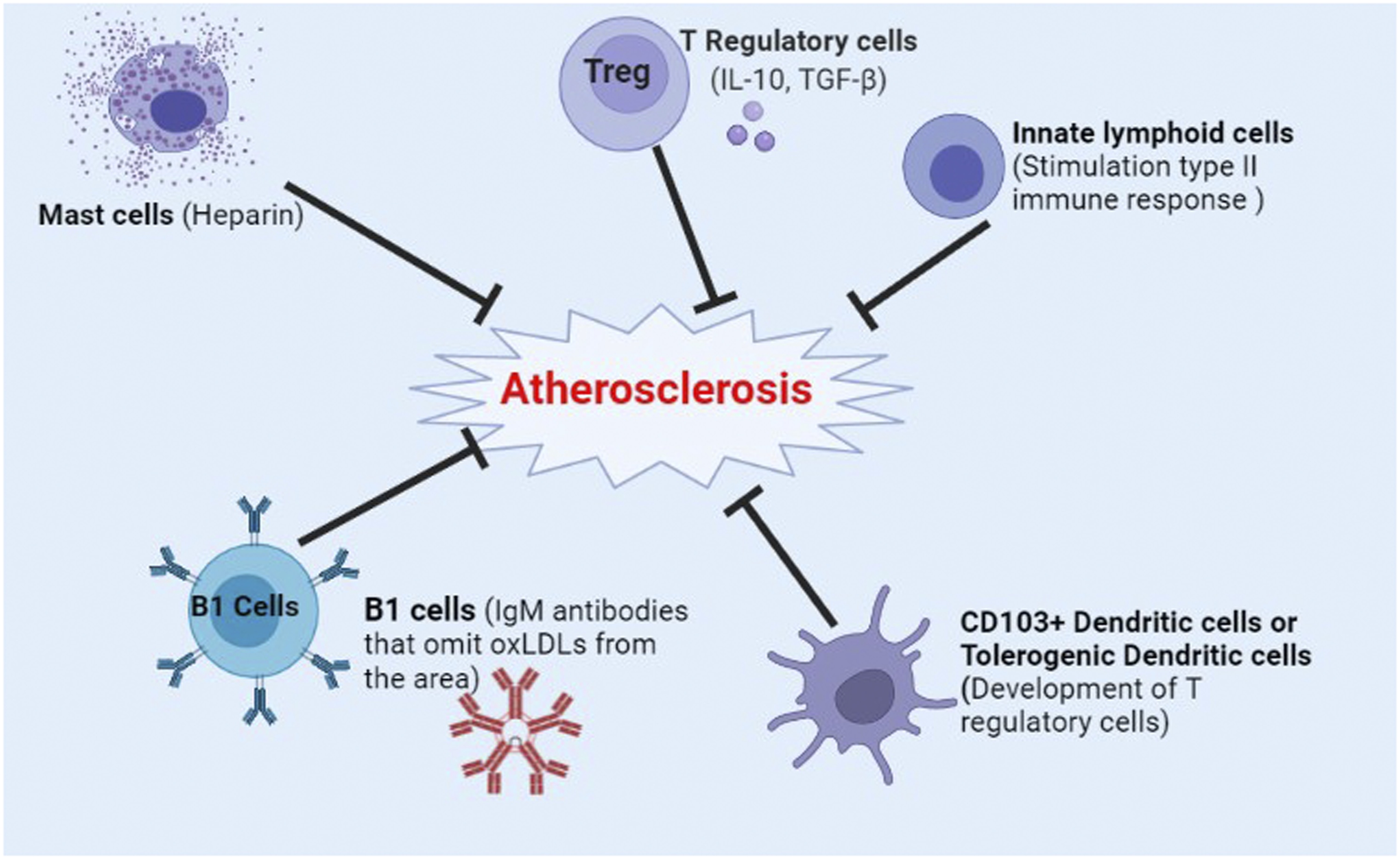
Components of the immune system that promote atherosclerosis. NKT cells, Natural killer T cells; IL-1, Interleukin 1; IL-4, Interleukin 4; IL-10, Interleukin 10; IL-12, Interleukin 12; IL-18, Interleukin 18; IFN-γ, Interferon gamma; TNF-α, Tumor necrosis factor alpha; Th1, T helper 1 cells; IgG, Immunoglobulin G; IgE, Immunoglobulin E; ILC-1, Innate lymphoid cells 1; ILC-3, Innate lymphoid cells 3. Components of the immune system that inhibit atherosclerosis. Treg, T regulatory cells; IL-10, Interleukin 10; TGF-β, Transforming growth factor beta; IgM, Immunoglobulin M; OxLDL, Oxidized low-density lipoprotein; CD103+ Dendritic cells, Cluster of differentiation 113 positive dendritic cells.
Immune cells in the atherosclerotic plaque
Almost all of the innate and adaptive immune cells are implicated in the atherosclerosis process.26,27 Macrophages are dominant in number, and other cells such as mast cells, T lymphocytes, and B lymphocytes are found in the atherosclerotic lesions.27,28 Intriguingly, localization of these cells throughout the plaque may vary, and some of them are accumulated more in certain areas. For instance, macrophages, mast cells, and T cells are mostly seen in the shoulder regions of the atherosclerotic plaque.29–31 Here, we discuss the functions of impactful immune cells in relation to the formation and development of atherosclerosis according to their importance in this process.
Macrophages
Circulating monocytes and consequently, macrophages are the first immune cells appeared in the atherosclerotic lesion. 32 Both monocytes and macrophages are involved in different stages of the atherosclerotic process including plaque formation, progression, and rupture. 26 Number of circulating monocytes and number of macrophages in the plaque are closely related. 33 Interestingly, type of receptors on circulating monocytes is associated to the size of the plaque. While TLR-2 is expressed more in patients with larger plaques, CD14, TLR-4, and myeloperoxidase are dominant in patients with smaller plaques. 34 This difference in receptor expression may demonstrate the involvement of different types of macrophages in different stages of plaque development. 34 Moreover, it was shown that monocytes with osteogenic markers are highly associated with atherosclerosis. 35 This may explain calcium deposition in the atherosclerotic plaques and vascular calcification in the coronary arteries of such patients. 36 Nonetheless, there is no relation between monocytes subpopulation and incidence of major adverse cardiovascular events in a 3-years follow up period. 37 However, an association is reported between nonclassical monocytes and CAD progression. In a recent study, Slan+ monocytes are shown to resolve plaques due to their anti-inflammatory property. The authors concluded that this subset helps to prevent atherosclerosis progression. 38 There is an association between increased number of plaque macrophages and plaque development. In early stages of plaque formation, monocytes depletion results in attenuation of atherosclerosis progress though preformed plaques are not influenced by monocytes count, 39 showing the more complex condition in the advanced stages. 26
There is a direct correlation between clinical cardiovascular risk factors and the amount of infiltrated macrophages within the plaque. 40 Although symptomatic plaques are infiltrated with more macrophages in comparison to asymptomatic ones, 41 marked macrophage infiltration does not necessarily translate into poor outcome. Lower risk of restenosis in 1-year follow up was reported in patients with higher content of plaque macrophages. However, this study reported the histological findings of atherosclerotic plaques obtained from carotid endarterectomy, which might be pathologically different from that of de novo atherosclerosis. 42 Of factors that precisely predict future cardiac events are plaque neovascularization and intraplaque hemorrhage. 43
The sources of monocytes are hematopoietic stem and progenitor cells (HSPCs) in the bone marrow. During atherosclerosis, HSPCs translocate to the spleen, and extramedullary hematopoiesis is performed to produce specific type of monocytes (lymphocyte antigen 6 complex, Ly6Chigh). 44 These splenic monocytes with inflammatory properties infiltrate into the atherosclerotic lesion. 44 Hyperlipidemia and low levels of HDL accelerate HSPCs proliferation and monocytosis, totally aggravating atherosclerosis progress. 45 Hyperglycemia imposed similar effect on the monocyte count via augmenting progenitors proliferation. 46 Intriguingly, splenic migration of HSPCs and generation of monocytes in the new site are intensified after myocardial infarction (MI). This inevitably escalates atherosclerosis progress, and increases the risk of further cardiovascular events. 47 Whereas entrance of splenic monocytes to the blood stream is mediated by angiotensin II 48 and splenic B cells, 49 blocking of this entrance ameliorates the MI adverse consequences. 48
Macrophages play a delicate role in orienting plaque fate. They affect growth and rupture of the plaque through either maintaining or attenuating inflammation. This is done via secretion of anti-inflammatory cytokines and burying dead cells by efferocytosis process. 26 Based on promotion or inhibition of inflammation, macrophages are classified into M1 and M2 types, respectively. 50 Some factors determine the macrophage polarization. Anatomical site of the plaque, carotid or femoral, affects M1/M2 proportion, with the former site contains higher M1. 51 M1 polarization strongly activates inflammatory pathways within the plaques. 52 Polarization state and macrophage phenotype are also influenced by a plethora of cytokines and growth factors in the atherosclerotic lesions. 53 However, categorization of macrophages into M1 and M2, and relating this classification to the atherosclerosis progress is not as simple as it seems because both types are tightly involved in lesion development. 54
In the microenvironment of human plaque, lesional macrophages possess specialized functions that are not only explained by M1 and M2 phenotypes. 55 A variety of macrophage populations with diverse functions are associated to the atherosclerosis. 56 Depending on the activation stimuli received by M2 macrophages, they undergo phenotypic changes to generate several subclasses including M2a, M2b, M2c, and M2d. 57 M2a is emerged after stimulation by IL-4 and IL-13, and secrete pro-fibrotic factors involved in tissue repair. M2a types are known as wound-healing macrophages.58–62 However, M2b macrophages, which are induced by IL-1β or LPS, are known for simultaneous production of anti-inflammatory and pro-inflammatory cytokines. 63 M2c macrophages are induced upon exposure to IL-10 and glucocorticoids, and together with M2b phenotypes are called regulatory macrophages. 64 The characteristics of M2d macrophages are releasing high levels of IL-10 and vascular endothelial growth factor besides low level of TNF and IL-12.65,66
Furthermore, there are other macrophage phenotypes that are plaque specific such as Mox, Mhem, and M4. 57 CXCL4 and oxLDL induce emergence of M4 and Mox macrophages, respectively. Both of them have atherogenic properties. After intraplaque hemorrhage and hemoglobin release which especially occurs in advanced lesions, another subtype called M (Hb) or Mhem is produced. 67 It is noteworthy that each phenotypic subset has a distinctive function in the course of atherosclerosis development.
Ly6Chigh monocytes contribute greatly to the generation of local macrophages. 68 Existence of proliferating macrophages is also evident in the atherosclerotic lesions.69,70 oxLDL propels macrophage proliferation, too.71,72 In advanced lesions, the frequency of macrophages (up to 87%) is mainly determined by local proliferation.73,74 Contrary findings showed that the rate of proliferation is higher in the early lesions. 75 In addition to the macrophages, proliferation of monocytes could occur locally at the lesion site. 76 Maturation of the macrophages and monocytes results in acquisition of phagocytic property. Macrophage colony-stimulating factor (MCSF) provokes differentiation of monocytes to macrophages in the endothelial layer. Upon formation of macrophages, their scavenger receptors mediate oxLDL internalization. 77 Macropinocytosis and TLRs-mediated signals are involved in this process. 78 Ingestion of LDL particles by the macrophages results in the accumulation of cytoplasmic lipid droplets. Meanwhile, cholesterol esters form a foamy shaped cell symbolized as the dawning of the atherosclerosis. Thereafter, fatty streaks, which are the typical histological finding of atherogenesis, emerged. 79 Recent studies revealed the possibility of lipid loading by circulating monocytes as well. They can also acquire a foamy phenotype, and infiltrate into the atherosclerotic lesion. 80 This emphasizes the high concentration of serum monocytes as a risk factor for atherosclerosis development. 81 Notably, it is not clear that which comes first to the plaque site, circulating monocytes or LDL particles. Possibly, both players enter to the subendothelial layer in parallel. 82
Cholesterol loading and transdifferentiation to macrophage-like cells may also be performed by SMCs. 83 Therefore, about 50% of the foam cells are reported to have SMC origin. 84 However, function and transcriptional profile of SMCs-derived foam cells are different compared with those originated from macrophages. The former has reduced potential of phagocytosis and efferocytosis.85–87 Interestingly, cholesterol unloading reverses the foamy phenotype to their original SMC. 26
Mast cells
Mast cells have been historically identified in the atherosclerotic lesions. 88 They are present in the adventitial layer of the artery, and are implicated in almost all of the steps from early to the late stages of atherosclerosis, with their counts increasing with lesion development. 19 Mast cells recruit neutrophils and circulating leukocytes to the plaque site. So, they contribute to the initiation of the plaque growth and mounting of the inflammatory responses. 19 This recruitment is performed following secretion of attractant chemokines like TNF-α and neutral proteases from activated mast cells. Mast cells are also involved in adhesion to the activated endothelial cells followed by transendothelial migration. 19 In fact, activated mast cells stimulate endothelial cells to express adhesion molecules.89,90 Indeed, mast cells secrete tryptase, which disrupts endothelial integrity impairing its barrier function. 19 LDL particles may be emerged after tryptase-induced degradation of the endothelium. 91 Chymase is the other enzyme that is secreted by the mast cells. It inhibits efflux of the cholesterol from the plaque back to the circulation, and may also decrease anti-inflammatory properties of HDL.92,93
Histamine is another important product of the mast cells. It increases microvascular permeability particularly in the venous side of the vessels through activation of endothelial histamine H1 receptor facilitating entrance of more LDL particles into the intima. This effect intensifies the progress of atherosclerosis.94,95 While histamine and leukotrienes of the mast cells are atherogenic,89,96 mast cell-derived heparin is likely to limit the growth of thrombus. 19 Pro-inflammatory cytokines with atherogenic characteristics such as IL-6 and IFN-γ are also secreted by the mast cells. 97 In contrary to heparin and tryptase, histamine is not a specific product of the mast cells, and is also synthesized by the endothelial cells. 89
Plaque growth is largely influenced by the proliferation of SMCs and deposition of extracellular matrix. Contribution of mast cells to the proliferation of pulmonary SMCs was carried out through secretion of mitogenic factors. 98 However, there is no data about such mast cell-derived effect on arterial SMCs. 19 Secretion of growth-limiting factors by the endothelial cells is attenuated upon endothelial erosion.99,100 This endothelial erosion that is supported by the mast cells probably provide the opportunity for the growth of subendothelial SMC.101,102
Neutral proteases and histamine release of the mast cells cause apoptosis of the macrophages within the lesion. 103 Apoptotic area is mainly located at the center of the plaque. When mast cells are activated locally, apoptotic cells are increased leading to the growth of apoptotic core and stabilization of the plaque. 19 In an opposing role, mast cells induce impairment and apoptosis of SMCs which result in weakening and rupture of the atherosclerotic plaque. 104 For instance, chymase degrades fibronectin leading to SMCs apoptosis. Fibronectin is a precellular component of matrix which is necessary for adhesion of SMCs and survival signaling. 105
Growing of the atherosclerotic plaque which is accompanied with narrowing of the lumen cause turbulence of the blood flow. Turbulence-induced stress makes endothelial cells dysfunctional, and even detach. Local mast cells may be activated on this occasion, and secret tryptase and chymase. These enzymes disintegrate the basement membrane aggravating separation of the endothelial cells. 100 TNF-α is released from the activated mast cells, and deteriorates survival signaling of the endothelial cells smoothing their apoptosis. 99 Also, circulating neutrophils which are recruited to the injured area through the effect of mast cell-derived IL-8 may contribute to the endothelial erosion.106,107 Activated mast cells besides activated neutrophils drive degradation and rupture of the plaque cap, from both abluminal and luminal sides, respectively. 89
Dendritic cells
Different types of dendritic cells (DCs) are perceived as important players in all stages of the atherosclerosis process. These cells have crucial roles in lipid uptake and lipid metabolism, and are considered as important mediators in early accumulation of lesional lipid. 108 In vivo studies demonstrated the induction of DC maturation by cholesterol crystals, inflammatory cytokines, and necrotic cells-derived nucleic acids in the atherosclerotic plaques. There is a strong link between DC maturation and atherosclerosis as DC maturation has two effects; secretion of pro-inflammatory cytokines and activation of T cells.109,110 CD103+ DCs are known as tolerogenic DCs that induce the development of T regulatory cells (Tregs). Tregs suppress activation of endothelial cells 111 and macrophages via secretion of anti-inflammatory cytokines such as TGF-β and IL-10112,113 leading to atheroprotection. In contrast, CCL17+ DCs are known as suppressors of Treg development since they act on CCR4, and promote apoptosis of the Tregs. 114 Plasmocytoid DCs (pDCs) have been detected in the shoulder regions of the atherosclerotic plaques, 115 and they are one of the sources of IFN-α and IFN-β, which demonstrate pro-atherogenic functions. 116 Both immature classical DCs and CD103+ DCs are potent efferocytes. Secondary cellular necrosis, which occurs in those apoptotic cells that were not ingested by the phagocytes, is considered as a pro-atherosclerotic process which is prevented by DC efferocytosis. 117 Also, DC mediated efferocytosis of lipid-laden apoptotic foam cells is an important mechanism for eliminating cholesterol, oxidized lipids, and other pro-inflammatory DAMPs.118–121
T lymphocytes
T lymphocytes, in particular Th cells (CD4+ T cells), infiltrate into the plaque and recognize protein antigens of oxLDLs, heat-shock proteins, and proteins of the pathogens like Chlamydia species.122–124 After activation, Th cells release IFN-γ, which in turn activates macrophages. In particular, Th1 cells enhance antigen presentation as well as help to synthesize cytokines such as TNF and interleukin 1. 125 In advanced atherosclerotic lesion, a large number of T cells, especially Th1, are present. This subtype, which is stimulated by IL-18 and IL-12, accelerates atherosclerosis. However, there are debates regarding the role of Th2 cells in the atherosclerosis development. 77 It seems that during atherosclerosis, Th1 response rather than Th2 is emerged due to signaling of certain cytokines within the plaque.126,127 Although it was reported that Th2 response may have antiatherosclerotic effect in some studies, 128 it imposes some unfavorable outcomes as well, 2 such as formation of aneurysms which is induced via Th2 pathway. 129
Cytotoxic T cells (CD8+ T cells), 130 Natural Killer T (NKT) cells, 1 NK cells 130 and γδ-T cells 131 are among the most important types of killer cells. Coronary plaques with few SMCs and large amount of lymphocytes have thin and vulnerable fibrous cap. The killer lymphocytes induce cell apoptosis in the lesions through different approaches like cytotoxin-, FasL/TRAIL- and/or cytokine-dependent mechanisms 132 that ultimately terminates in plaque rupture and sudden death. 133 Along with CD4+ T cells, killer lymphocytes are all implicated in the atherosclerosis process. They are copious in the unstable plaques reinforcing their role in plaque rupture as well as their involvement in the initial development of atherosclerosis. 132
Killer cells could also be categorized into innate or adaptive ones based on the type of activation. 132 Nonetheless, cytotoxic potential of both types causes cell death resulting in plaque rupture. However, time and mechanism of their implication to form vulnerable plaques are poorly understood. Also, it is yet unclear whether they are atherogenic per se or their atherogenicity is derived indirectly from interaction with other cell types. 132
NKT cells are in fact lipid-sensing cells. 18 They secret pro-atherogenic cytokines and cytotoxins. 18 Unique T cell receptors (TCRs) along with common surface markers as with NK cells are expressed by NKT cells. 18 iNKT cells have roles in the initiation and development of atherosclerotic lesions. The effects of NKT cells in atherosclerosis are delivered in the form of secretion of certain cytokines like IFN-γ, IL-4 and IL-10 supplemented by cytotoxic potential of NKT cells after activation. Also, these cells affect the stability of the lesions by secretion of granzyme B and perforin, which are both involved in the formation of necrotic core. 18 In contrary, natural and induced regulatory T cells are known as atheroprotective T cells because they diminish inflammatory responses or deactivate dendritic cells. 134
B lymphocytes
B cells in the lesions, although rare in quantity, show antiatherosclerotic activity possibly due to the production of cytokines and antibodies against plaque antigens as well as binding of antibodies to Fc receptors. Antibodies of splenic B cells recognize phosphorylcholine molecule in oxLDLs and apoptotic cell membranes. Therefore, they effectively inhibit atherosclerosis through excluding oxLDLs from further involvement in the atherosclerotic process.135,136 The notion of atheroprotectivity of B cells originates from studies in which atherosclerosis is exacerbated after complete or splenic B cell deficiency.135,137,138
Although secretion of antibodies by B cells, as a fundamental part of humoral immunity, has an atheroprotective effect, different types of B cells (B1 and B2) are not similar regarding the effect on the atherosclerosis development. B1 cells produce IgM antibodies and possess an atheroprotective function through omitting oxLDLs from the environment 139 while B2 cell secret IgG and IgE which promote atherosclerosis. 77 However, there are controversies in this regard. Low circulating IgM levels are associated with extensive atherosclerosis and cardiovascular events. Intriguingly, the relationship between IgG levels and atherosclerosis development could have been inverse, direct, or neutral. 140 Also, it was shown that severity of CAD and coronary events are increased in the case of elevated serum IgE. 141 In addition to the humoral immune responses, B cells are also involved in atherosclerosis in the form of cellular immunity. This involvement is a complex interaction between T and B cells that include, but not limited to, regulation of T cell activation via antigen presentation, cytokine production, and co-stimulation. 27
Secondary lymphoid organs are involved in the atherosclerosis-related immune responses by the intermediary of B cells. 27 Although number of B cells in the atherosclerotic plaques are few, they are abundant in artery tertiary lymphoid organs. 142 These are aggregations of B cells, T cells, and plasma cells in the adventitia layer of advanced atherosclerotic plaques with potential to mount local immune responses. 27
Innate lymphoid cells (ILC)
A recently discovered population of immune cells is known as innate lymphoid cells. They affect other cells via two main mechanisms: first, secretion of cytokines and other soluble factors, second, cytotoxic function. These cells stimulate type II immune response, and attenuate the atherosclerosis process. 143 Similar to other immune cells, innate lymphoid cells have several subtypes. Cytokines released from ILC-1 and ILC-3 subtypes are involved in the progression of atherosclerosis. 143
Platelets and neutrophils
Activated platelets release certain chemokines such as CCL5 that causes recruitment of neutrophils to the atherosclerotic lesions through CCR1 and CCR5. 144 Neutrophils secrete granule proteins like azurocidin, cathepsin G, and α-defensins. They are involved in activating macrophages and recruiting monocytes to the injured site. Totally, these movements promote formation of the foam cells. 144 In the conditions of chronic inflammation like atherosclerosis, destructing function of the neutrophils may divert from foreign cells toward self-cells, which might lead to the vascular injury. 145 However, type of vascular injury could be variable in different stages of the atherosclerosis progress. While in the early stages of atherosclerosis, neutrophils cause endothelial dysfunction and vascular inflammation, they provoke some events like plaque rupture and atherothrombosis at the late stages. 145 Defensins and cathelicidins are neutrophil peptides with prominent roles in the innate immunity. High concentrations of these two molecules are detectable in the atherosclerotic plaques. In fact, they are mediators of vascular disease. Defensins instigate the metabolism of vessel walls lipoprotein besides accumulation and modification of LDL and lipoprotein in the endothelium and extracellular matrix. Defensins are prothrombotic elements that interfere with the function of vascular SMCs, too and, inhibit angiogenesis as well. Cathelicidins enhance in vitro endothelial proliferation, induce angiogenesis, and regulate apoptosis of the endothelial cells. 146
Neutrophils paly a substantial role in the atherosclerosis by another mechanism, too. Neutrophil extracellular traps (NETs) are mesh-like projections with different constituents which are released after neutrophil activation. NETs extrusions are involved in a variety of pathological conditions including atherosclerosis and thrombosis. 147 NETs contain decondensed chromatin, histones, other nuclear and cellular proteins, cytoskeleton, proteases, and azurophilic granules. They also harbor some circulating elements like tissue factor, fibrin, and other similar proteins that are important in coagulation. 148 NETs induce oxidative stress and oxidize HDL. They cause dysfunction of the endothelial cells followed by apoptosis, totally give rise to the generation of anti-double-stranded-DNA autoantibodies. 147
NETs are found in the atherosclerotic lesions and arterial thrombi. They activate endothelial cells, antigen presenting cells, and platelets leading to pro-inflammatory immune reactions. Accordingly, their presence in the components of atherosclerosis process is dynamic. 149 NETs are capable to trigger coagulation toward formation of atherosclerotic plaques. 150 They contribute significantly to atherogenesis as NETs facilitate the formation of a fibrin-like structure for adhesion, activation, and aggregation of platelets. Moreover, they encourage accumulation of von Willebrand factor and fibrinogen, two prothrombotic molecules, resulting in the formation of thrombosis. They are associated to arterial and venous thrombosis which were reported in both humans and animal models. 147
Limitations
The present study suffers from inherent limitations subjected to the review articles including the effects of authors’ personal view, and unintentional errors in data translation from original studies in terms of mispresentation or misinterpretation. Specifically, the present review discusses major players of the immune system involved in the initiation and development of atherosclerosis. However, there are still uncovered aspects or those that are not yet discovered, which need further investigations. More importantly, the principle starter of the atherosclerosis process is still not fully elucidated. Is such a factor present at all? Is it present at the lesion site or act remotely? Is this a common factor between patients with atherosclerotic plaques? Is it feasible to target this basic factor in order to stop the growing trend of morbidity and mortality?
Conclusion
Atherosclerosis has become the affliction of human kind in the modern world. There is an essential need to have an in-depth understanding about the effectors of its initiation and development. Immune system and its components make fundamental contribution to the atherosclerosis process. They play crucial roles in different stages of the plaque progression. Elucidation of the mechanisms used by these immune cells helps to apply efficient treatment, and even preventive strategies.
Footnotes
Author contributions
Iman Razeghian-Jahromi: Conceptualization, writing the original draft, review & editing. Ali Karimi Akhormeh: Writing the original draft & review. Mohammad Javad Zibaeenezhad and Mahboobeh Razmkhah: Writing the review & editing. All authors read the manuscript critically, and approved it.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported by Shiraz University of Medical Sciences [grant number: 24943].