
Abstract
Background
Ovarian cancer is a lethal malignancy with limited therapeutic options, highlighting the urgent need to identify reliable biomarkers and therapeutic targets. This study investigates the clinical and oncogenic significance of β-hydroxybutyrate dehydrogenase 1 (BDH1) as a potential therapeutic target in ovarian cancer.
Methods
Integrative genomic analyses were conducted to evaluate the oncogenic potential of BDH1 by assessing its gene amplification and expression patterns in ovarian cancer. Functional assays, including BDH1 knockdown, were performed to examine its effects on proliferation, colony formation, cell cycle, and stemness. Mechanism exploration involved assessing the Wnt/β-catenin pathway. Virtual screening was used to identify BDH1 inhibitors.
Results
BDH1 was identified as a crucial oncogene in ovarian cancer progression, characterized by frequent gene amplification and overexpression, which strongly correlated with advanced disease stage and poor patient prognosis. Functionally, BDH1 promoted tumor progression by regulating G1/S transition proteins and maintaining cancer stemness via Wnt/β-catenin signaling. BDH1 depletion suppressed malignant phenotypes, confirming its oncogenic role. BDH1 was pharmacologically targeted by centrinone, which effectively downregulated oncogenic effectors.
Conclusion
Our study underscores BDH1 as a multifunctional oncogenic driver in ovarian cancer. The strong association of BDH1 with aggressive disease underscores its clinical relevance as a prognostic predictor, while its functional role and pharmacological inhibition position BDH1 as a promising therapeutic target for ovarian cancer.
Introduction
Ovarian cancer (OV) is one of the most lethal gynecological malignancies, with a high mortality rate due to its asymptomatic early stages and frequent diagnosis at advanced stages.1,2 Epidemiological studies indicate that OV accounts for approximately 3% of all cancers in women, with over 300,000 new cases diagnosed annually worldwide. 3 The majority of OV cases are epithelial ovarian cancers, which are further classified into serous, endometrioid, mucinous, and clear cell subtypes, with high-grade serous ovarian cancer being the most aggressive and prevalent form. 4 Despite advances in surgical techniques and chemotherapy, the 5-year survival rate for advanced-stage OV remains below 30%, underscoring the urgent need for novel therapeutic targets and biomarkers to improve patient outcomes.5,6 The tumor microenvironment (TME) of OV, characterized by immune evasion, stromal interactions, and metabolic reprogramming, plays a critical role in disease progression and therapy resistance, making it a focal point for current research. 7
The BDH1 gene encodes for β-hydroxybutyrate dehydrogenase 1, a mitochondrial enzyme traditionally known for its role in ketone body metabolism, catalyzing the interconversion of β-hydroxybutyrate and acetoacetate. 8 Beyond its metabolic functions, emerging evidence suggests that BDH1 is implicated in various cancers, including breast, 9 liver, 10 prostate, 11 and lung cancer, 12 where it contributes to tumor growth, metastasis, and chemoresistance. In OV, BDH1 has been linked to poor prognosis, but its precise mechanisms remain poorly understood. 13 Recent studies highlight its potential involvement in cell cycle regulation, stemness maintenance, and activation of oncogenic signaling pathways, such as PI3K-AKT and Wnt.10,14 These pathways are critical for cancer cell proliferation, survival, and differentiation, suggesting that BDH1 may serve as a central node in OV pathogenesis. However, a comprehensive investigation into the functional roles and clinical significance of BDH1 in OV is still lacking, particularly its impact on the TME and cancer stemness.
Cancer stem cells (CSCs) are a subpopulation of tumor cells with self-renewal capacity and resistance to conventional therapies, driving tumor recurrence and metastasis.15,16 In OV, CSCs are enriched in the TME and contribute to disease progression through mechanisms involving metabolic flexibility and signaling pathway activation. 17 The Wnt/β-catenin pathway is a key regulator of CSC population maintenance and has been implicated in OV aggressiveness.18,19 Given the reported associations between BDH1 and stemness-related processes in other cancers, it is plausible that BDH1 may similarly influence CSC properties in OV. 10 Furthermore, metabolic reprogramming is a hallmark of cancer, and enzymes like BDH1, which bridge metabolic and signaling networks, represent promising therapeutic targets.
In this study, we aimed to elucidate the clinical and functional significance of BDH1 in OV, leveraging multi-omics data from the TCGA-OV cohort and single-cell RNA sequencing (scRNA-seq) to explore its expression patterns and associations with tumor progression. We further investigated the mechanistic roles of BDH1 in cell cycle regulation, stemness acquisition, and Wnt pathway activation using in vitro models. By integrating clinical, molecular, and pharmacological analyses, our study provides a comprehensive understanding of BDH1 in OV pathogenesis. The insights gained not only advanced mechanistic knowledge of OV progression but also paved the way for developing targeted therapies aimed at disrupting BDH1-mediated oncogenic networks.
Method
Cell culture and sgRNA-mediated transfection
The human OV cell line SKOV3 was obtained from the American Type Culture Collection and cultured in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco) at 37°C in a humidified 5% CO₂ incubator. For BDH1 knockdown, two independent signal RNAs (sgRNAs) targeting BDH1 (sgBDH1-1: 5ʹ-CATAAGTCCGACGGCCAATC −3ʹ; sgBDH1-2: 5ʹ-GATTGTCCGCTCGAGCCTGA −3ʹ) and a non-targeting control sgRNA were adopted. Transfection was conducted using Lipofectamine™ 3000 (Invitrogen) according to the manufacturer's protocol. Cells were cultured for 48 h following transfection with Lipofectamine 3000 before functional assays. The knockdown efficiency was validated by real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) and Western blot.
MTT assay
Cell viability was assessed using the MTT assay. Briefly, 5 × 103 cells/well were seeded in 96-well plates and treated with shBDH1/varying concentrations of centrinone (0–500 nM) or DMSO control for 48 h. After treatment, 20 μL of MTT solution (5 mg/mL; Sigma) was added to each well and incubated for 4 h at 37°C. The formazan crystals were dissolved in 150 μL DMSO, and absorbance was measured at 570 nm using a microplate reader (BioTek). For the time-course assay, cell viability was measured at 24, 48, 72, 96, 120, and 144 h post-treatment. The IC50 value for centrinone was calculated based on the 72-h time point and calculated using GraphPad Prism.
Colony formation assay
SKOV3 cells (500 cells/well) were seeded in 6-well plates and cultured for 10–14 days. Colonies were fixed with 4% paraformaldehyde (PFA) for 15 min, stained with 0.1% crystal violet (Sigma) for 30 min, and washed with phosphate-buffered saline. Colonies were counted manually. Colonies were allowed to form over a period of 14 days post-seeding, with medium refreshed every 3 days, before fixation and staining.
qRT-PCR
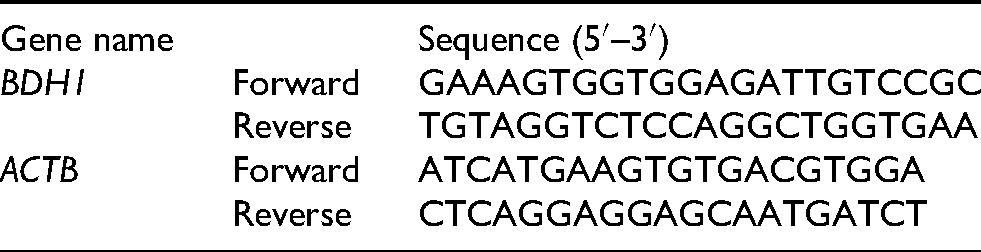
Total RNA was extracted from cultured cells 48 h post-transfection using TRIzol reagent (Invitrogen) following the manufacturer's protocol. RNA concentration and purity were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific), with A260/A280 ratios between 1.8 and 2.0 considered acceptable. PrimeScript RT Master Mix (Takara) was used for complementary DNA synthesis. qPCR was performed using TB Green Premix Ex Taq (Takara) according to the manufacturer's instructions. Relative gene expression was calculated using the 2−ΔΔCt method, with ACTB as the internal control. The sequences of primers for each gene were as follows:
Western blot
Protein lysates were collected 48 h post-transfection using RIPA buffer (Thermo Fisher) supplemented with protease and phosphatase inhibitors (Roche). Protein concentrations were determined using the BCA assay (Pierce). Equal amounts of protein (30 μg/lane) were separated by 10% SDS-PAGE and transferred to PVDF membranes (Millipore). Membranes were blocked with 5% non-fat milk in TBST for 1 h and incubated overnight at 4°C with corresponding primary antibodies. After incubation with HRP-conjugated secondary antibodies (1:5000, Dako, cat. no. P0260), protein bands were visualized using ECL substrate (Pierce) and quantified with ImageJ. The primary information of antibodies adopted in this work were listed below: BDH1 (1:1000; Proteintech, cat. no. 67448-1-Ig), pRb (1:1000; Cell Signaling Technology (CST), cat. no. #9308), CDK4 (1:1000; CST, cat. no. #12790), p21 (1:1000; CST, cat. no. #2947), p27 (1:1000; CST, cat. no. #3688), active-β-catenin (1:1000; CST, cat. no. #8814), Nanog (1:1000; CST, cat. no. #4903), SOX2 (1:1000; CST, cat. no. #23064), KLF4 (1:1000; CST, cat. no. #51221), and β-actin (1:2000; CST, cat. no. #8457).
Immunofluorescence
Cells grown on coverslips were fixed 48 h post-transfection with 4% PFA for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 5% bovine serum albumin for 1 h. Cells were incubated overnight at 4°C with anti-active-β-catenin antibody (1:50; CST, cat no. #8814), followed by Alexa Fluor 488-conjugated secondary antibody (1:500; Invitrogen) for 1 h. Nuclei were counterstained with DAPI (Sigma). Images were acquired using a confocal microscope (Zeiss LSM 880) and analyzed with ZEN software.
Public database-based bioinformatic analysis
The Cancer Genome Atlas (TCGA)-OV data illustrating the expression and mutation features of ovarian serous cystadenocarcinoma patients were downloaded from the UCSC Xena database (https://xena.ucsc.edu/). Gene amplification and messenger RNA (mRNA) expression (FPKM values) of BDH1 were analyzed using cBioPortal (https://www.cbioportal.org/). Survival analysis was performed using R package “Survival.” The BDH1 high (BDH1+) cases were defined as the 10% samples (n = 31) with the highest BDH1 expression, while the BDH1 low (BDH1−) cases were defined as the 10% samples (n = 31) with the lowest BDH1 expression. Gene expression values (FPKM) were log₂-transformed and median-centered prior to analysis. Differently expressed genes (DEGs) were identified by R package “DESeq2” with the criteria of | log2(Fold Change) | > 1 and P-value < 0.05. Pathway enrichment analysis and Gene Set Enrichment Analysis (GSEA) was conducted using R package “ClusterProfiler” based on the Gene Oncology terms and KEGG pathways.
scRNA-Seq analysis
Raw scRNA-seq data (GSE184880) 20 were processed using Seurat (v4.0). Cells with < 200 or > 6000 detected genes or > 10% mitochondrial reads were filtered out. Data were normalized using the LogNormalize method, and highly variable genes were identified for principal component analyses (PCA). Normalization was performed using the LogNormalize method with a scale factor of 10,000, followed by identification of the top 2000 highly variable features using the “vst” method in Seurat. Dimensionality reduction was conducted via PCA, where the first 30 principal components were selected for downstream analysis based on an elbow plot inspection. Uniform manifold approximation and projection (UMAP) was performed for nonlinear visualization using the RunUMAP function in Seurat with default parameters, a minimum distance of 0.3, and 30 neighbors. Cell clustering was carried out using the FindNeighbors and FindClusters functions, with a resolution parameter of 0.5 to achieve biologically meaningful cluster separation. InferCNV was adopted for the identification of malignant clusters. Immune cells from the same dataset as the reference baseline, and cells classified as aneuploid were annotated as malignant. Pseudotime analysis was performed with Monocle3 (v1.0). GSVACell clusters were annotated according to the markers listed below: T cell (CD3D, CD3E, CD8A, CD4); epithelial cell (KRT18, KRT19, EPCAM, CD24); monocyte (CD14, C1QA, VCAM); endothelial cell (PECAM1, CLDN5); cell cycle cell (MKI67, TOP2A); fibroblast (DCN, OGN); B cell/plasma (CD79A, JCHAIN), smooth muscle cell (SMC)/myofibroblast (ACTA2, MYH11, TAGLN). 20 Gene Set Variation Analysis (GSVA) was adopted to evaluate pathway activity variations across individual cells within the single-cell RNA-seq dataset, with enrichment scores (area under the curve; AUC) calculated for each cell based on predefined gene sets.
Virtual screening
The crystal structure of BDH1 was predicted by AlphaFold and downloaded under the accession number AF-Q02338-F1. was prepared using AutoDock Tools (v1.5.6). A library of 4485 US Food and Drug Administration-approved drugs (ZINC15 database) was docked using AutoDock4 (v4.2.1). The visualization of the binding conformation was conducted by PyMol.
Statistical analysis
Comparisons between two groups were analyzed using two-tailed Student's t-test. Survival differences were assessed by log-rank test. Spearman's correlation analysis was conducted by R package “stats.” P < 0.05 was considered statistically significant. All analyses were performed using GraphPad Prism 8.0.
Results
BDH1 exhibits frequent gene amplification and overexpression in OV, correlating with poor prognosis
In TCGA-OV cohort, BDH1 exhibited considerable amount of gene amplification in 45 out of 316 ovarian serous cystadenocarcinoma samples. Also, 21 cases demonstrated upregulated BDH1 mRNA expression level, while only 6 cases showed a downregulated BDH1 expression (Figure 1(a)). Analysis of somatic mutation data from the TCGA-OV cohort revealed that BDH1 is rarely mutated in OV, with only a single p.G26 V missense mutation identified at low allele frequency. This suggests that genomic amplification and transcriptional overexpression, rather than protein-altering mutations, represent the principal mechanisms of BDH1 dysregulation in this malignancy. In the scRNA-seq dataset, cells within OV TME were sub-grouped into eight specific cell types, namely T cell, monocyte, epithelia, B cell/plasma cell, cell-cycle cell, endothelial cell, fibroblast, SMC/myo-fibroblast (Figure 1(b)). BDH1 positive cells were enriched in the tumor samples (Figure 1(c)). Meanwhile, detailed analysis into the cell types in OV TME demonstrated a significant enrichment of BDH1 positive cells in the epithelia subcluster, indicating the upregulation of BDH1 mainly occurred in the cancer cells (Figure 1(d)). To add up with, the high expression of BDH1 was enriched in the late stage of OV progression (Figure 1(e)). As for the clinical significance, high expression of BDH1 predicted a remarkably poorer prognosis in OV patients (P = 0.0032) (Figure 1(f)). More specifically, the upregulation of BDH1 could serve as a poor prognosis predictor in late-stage OV patients (P = 0.012) (Figure 1(g)).
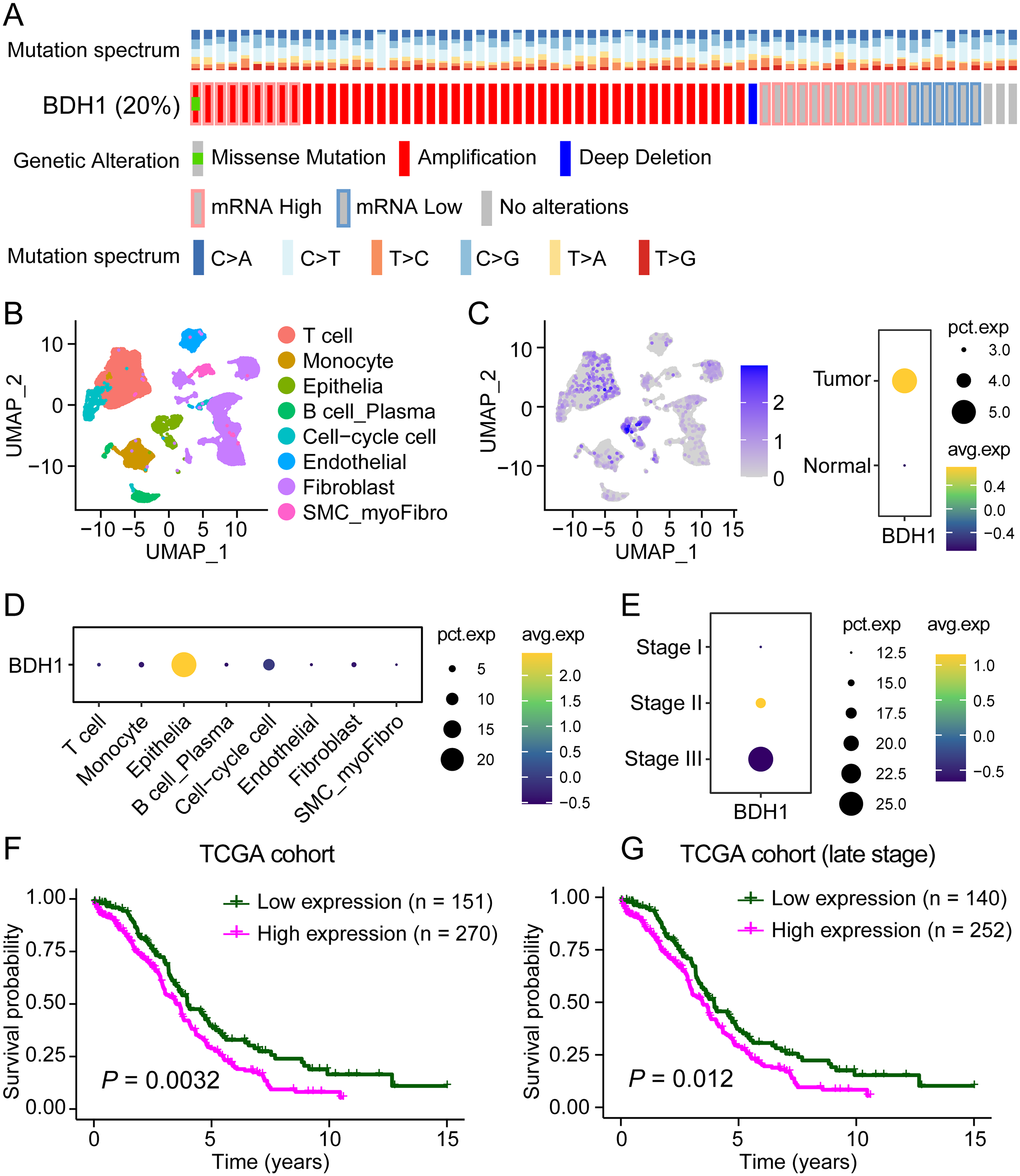
BDH1 demonstrates gene amplification and upregulated mRNA level in OV patients and predicts poor prognosis. (a) The copy number variation of BDH1 in TCGA-OV cohort. (b) Classification of all cell types within OV TME. (c) BDH1 expression was enriched in tumor samples. (d) BDH1 was specifically upregulated in epithelial cells. (e) BDH1 positive cells were enriched in late-stage OV samples. (f) High expression of BDH1 predicted poorer prognosis in OV patients. (e) BDH1 upregulation indicated worse clinical outcomes in late-stage OV patients.
BDH1 depletion suppresses malignant phenotypes in OV cells
The efficiency of two sgRNAs targeting BDH1 was firstly evaluated by RT-qPCR assays, and the results demonstrated that both significantly downregulated the expression level of BDH1 (Figure 2(a)). Afterwards, the MTT assay demonstrated that knocking down of BDH1 downregulated the cell viability of SKOV3 (Figure 2(b)). Also, the colony formation ability of SKOV3 was also downregulated by BDH1-depletion (Figure 2(c)). Pseudotime analysis revealed that BDH1 expression was predominantly enriched in cancer cells at early stages of the cell cycle (Figure 2(d) and (e)). The upregulation of BDH1 was chronologically correlated with proliferation-related markers, such as CDK1, CDC23, and CDC45, whereas apoptosis-related markers were elevated at the opposite end of the pseudotime trajectory (Figure 2(f)). Differential expression analysis was performed by comparing the top 10% (n = 31) and bottom 10% (n = 31) of samples based on BDH1 mRNA expression levels (FPKM values), as described in the Methods section. This analysis identified 1037 genes that were significantly upregulated in BDH1-high samples and 510 genes that were downregulated, based on the criteria of | log2(fold change) | > 1 and P-value < 0.05 (Figure 2(g)). Further enrichment analysis revealed that the cell cycling- and proliferation-related biological processes were enriched in the BDH1 + sample, such as epithelial cell proliferation, chromatin assembly, fibroblast proliferation, and regulation of DNA-binding (Figure 2(h)). The pathway enrichment analysis highlighted PI3K-AKT signaling pathway, TGF-β signaling pathway, Wnt signaling pathway, and Ras signaling pathway as the most upregulated pathways in the BDH1 + samples (Figure 2(i)).
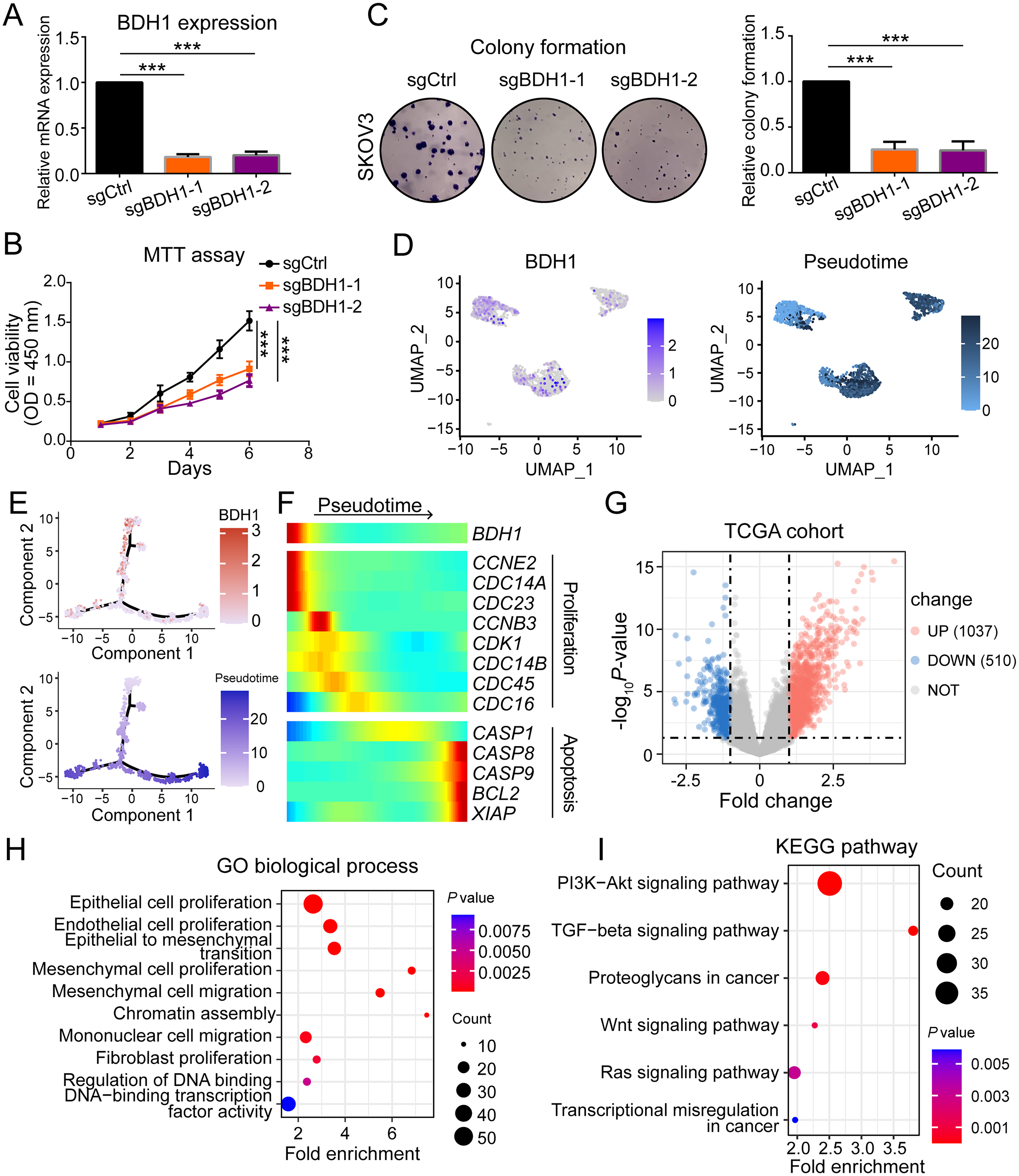
BDH1 is upregulated during early-stage OV cell cycling and promoted malignant features. (a) Evaluation of the knockdown efficiency of sgRNAs. (b) sgRNA-mediated knockdown of BDH1 downregulated the cell viability of SKOV3. (c) Knocking down of BDH1 impaired the colony formation ability of SKOV3 cells. (d) and (e) BDH1 upregulation was preferentially enriched in cancer cells during early cell cycle phases. (f) BDH1 upregulation was chronologically correlated with proliferation-related markers. (g) Volcano plot showing differentially expressed genes between BDH1-high (top 10%, n = 31) and BDH1-low (bottom 10%, n = 31) samples from the TCGA-OV cohort. Gene expression values (FPKM) were log2-transformed and median-centered prior to analysis. (i) KEGG pathways which were upregulated in the BDH1+ OV samples.
BDH1 expression correlates with cell cycle progression
The GSVA results revealed upregulated activities of three cell cycling-related biological processes, namely cell cycle, cell division, and DNA replication, in BDH1 positive cancer cells (Figure 3(a)). A detailed correlation analysis proved that the mRNA expression level of BDH1 was positively correlated with multiple cell cycling regulators in the TCGA cohort, including CDK1, CDK4, CCNB1, CCNB2, CDC6, CDC45, CDC25A, and CDC25B (Figure 3(b)). Furthermore, BDH1 + samples exhibited upregulated mRNA expression of multiple cell division cycle family proteins (CDCs), cyclin regulators (CCNs), and cyclin dependent kinases (CDKs). (Figure 3(c)). Western blot analysis revealed that BDH1 knockdown in SKOV3 cells led to downregulation of pRb and CDK4 concomitant with upregulation of p21 and p27, indicating BDH1's role in promoting cell cycle progression through modulation of G1/S checkpoint regulators (Figure 3(d)).
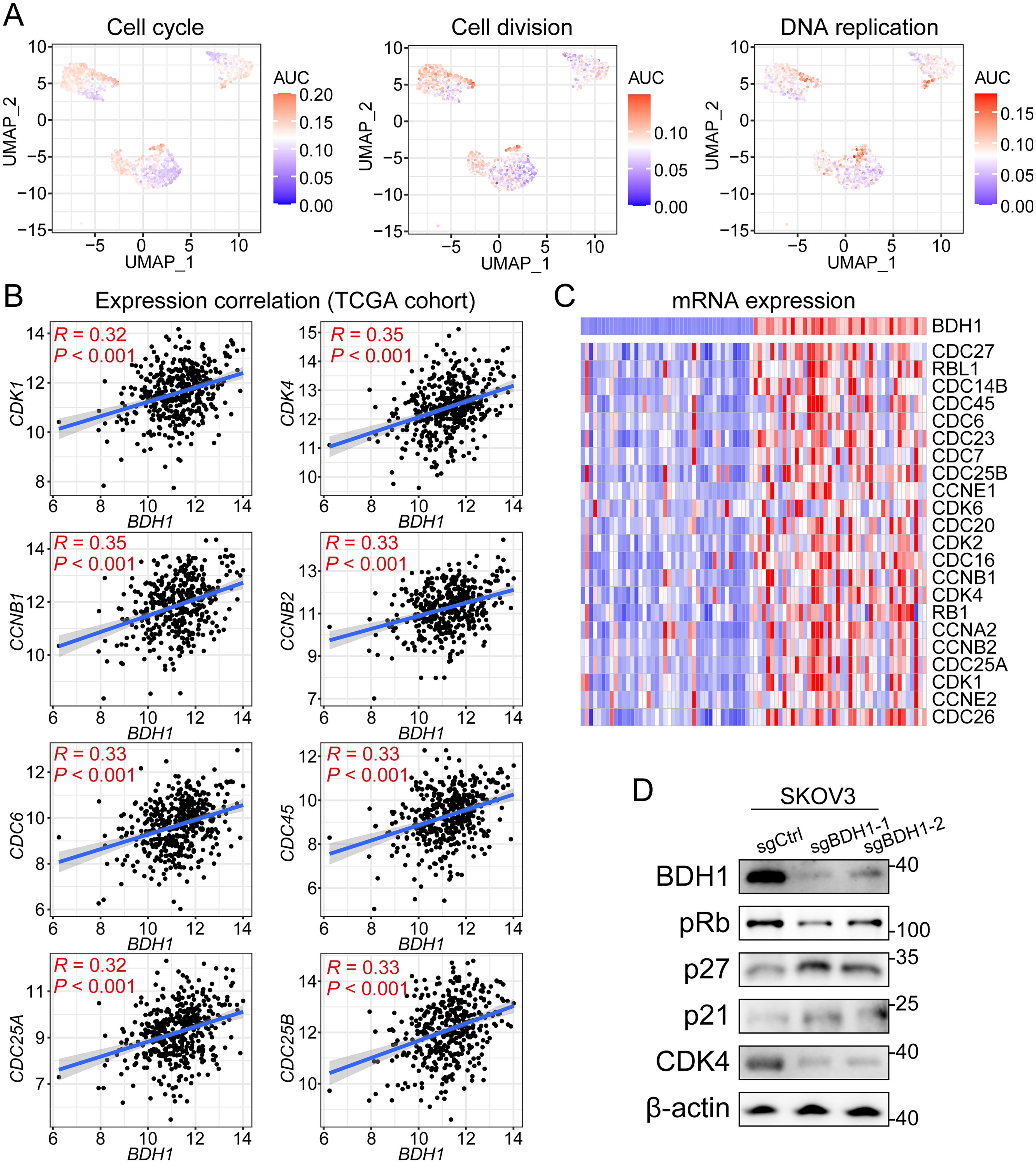
BDH1 regulates the cell cycling process of OV cancer cells. (a) GSVA enrichment analysis on biological processes across malignant cell clusters in scRNA-seq transcriptional atlas. (b) Correlation analysis between BDH1 and multiple cell-cycling regulators. (c) The mRNA expression pattern of various CDCs, CDKs, and CCNs in BDH1 + and BDH1- samples.
BDH1 maintains cancer stem cell properties in OV
CytoTRACE analysis revealed that BDH1 + cancer cells exhibited lower differentiation scores, suggesting enhanced stem-like properties (Figure 4(a) and (b)). Similarly, the GSVA results demonstrated relatively higher enrichment score in several cancer cell stemness-related biological processes, including stem cell proliferation, stem cell fate commitment, and stem cell population maintenance (Figure 4(c)). The GSEA was then conducted based on the DEGs between BDH1 + and BDH1- samples in TCGA-OV cohort. And the results demonstrated significant activation of stem cell development, stem cell differentiation, and stem cell proliferation processes in the BDH1 + samples (Figure 4(d)). To add up with, the pathway enrichment analysis also highlighted similar pathways that were hyper-activated in the BDH1 + samples (Figure 4(e)). The mRNA expression patterns of multiple stemness markers were visualized in the heatmap, which demonstrated significant upregulation in the BDH1 + samples when compared with the BDH1- samples (Figure 4(f)). The spearman's correlation analysis revealed positive correlation between the expression levels of BDH1 and four stemness-associated genes, namely EPCAM, POU5F1, LETM1, and FUT4 (Figure 4(g)). The regulatory function of BDH1 on cancer cell stemness acquisition was further validated by the Western blot assay, which exhibited downregulated expression of stemness marker CD44, Nanog, SOX2, and KLF4 in the BDH1-depleted SKOV3 cells (Figure 4(h)).
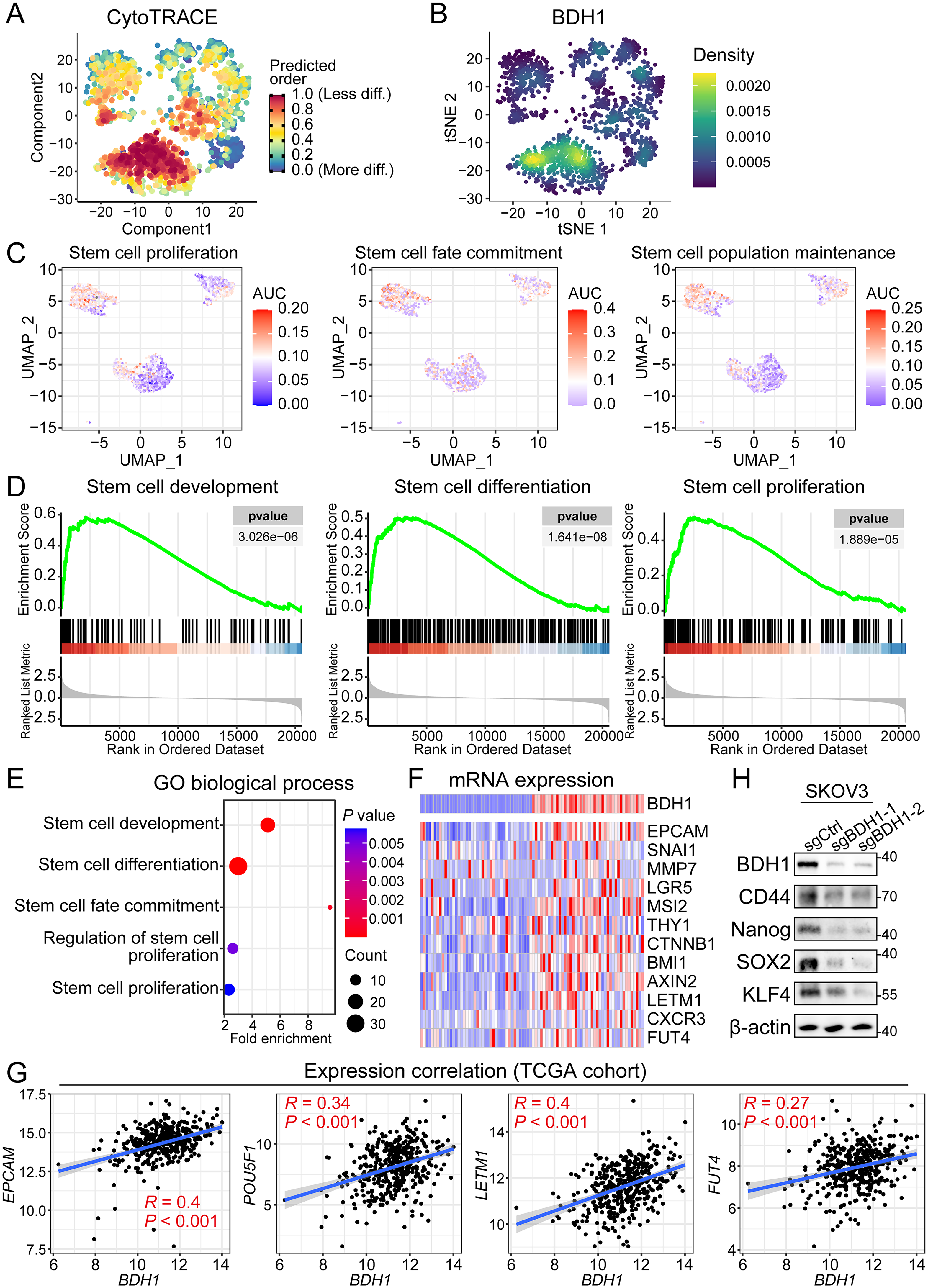
BDH1 mediates the stemness acquisition and CSC population maintenance during OV progression. (a) CytoTRACE analysis on the cancer cells in scRNA-Seq atlas revealing the predicted differentiation stage of cancer cells. (b) Density plot of the BDH1 + cells in tSNE dimension. (c) GSVA enrichment scores of stemness-related biological processes, including stem cell proliferation, stem cell fate commitment, and stem cell population maintenance across malignant cell clusters in scRNA-Seq transcriptional atlas. (d) GSEA results revealed significant hyper-activation of stemness-related pathways in BDH1 + samples, including stem cell development, differentiation, and proliferation. (e) Enrichment analysis demonstrated remarkable enrichment of stemness-related biological processes in BDH1 + samples, such as stem cell development, differentiation, and fate commitment. (f) Expression profile of cancer stem cell markers among BDH1 + and BDH1- samples. (g) Spearman's correlation analysis between the expression level of BDH1 and cancer cell stemness-related genes. (h) Western blot assay demonstrated downregulated expression of CD44, Nanog, SOX2, and KLF4 in BDH1-depleted cells.
BDH1 activates Wnt/β-catenin signaling to promote malignant phenotypes
The Wnt pathway was prioritized for subsequent analysis based on its well-characterized function in cancer stem cell maintenance and significant co-expression pattern with BDH1. The GSVA results demonstrated that Wnt signaling pathway was hyper-activated in the BDH1 + malignant cells among OV TME (Figure 5(a)). The expression correlation analysis revealed positive correlations between the expression level of BDH1 and CTNNB1, AXIN2, and MYC (Figure 5(b)). Meanwhile, the activation of Wnt signaling pathway was significantly enriched in BDH1+ OV samples, which was indicated by the GSEA results (Figure 5(c)). The immunofluorescence data revealed a decreased nuclear accumulation of active-β-catenin in SKOV3 cells after knocking down of BDH1 (Figure 5(d)). As for the downstream effectors, the expression level of cyclin D1 and c-Myc were both significantly downregulated by BDH1-depletion at both mRNA (Figure 5(e)) and protein level (Figure 5(f)).
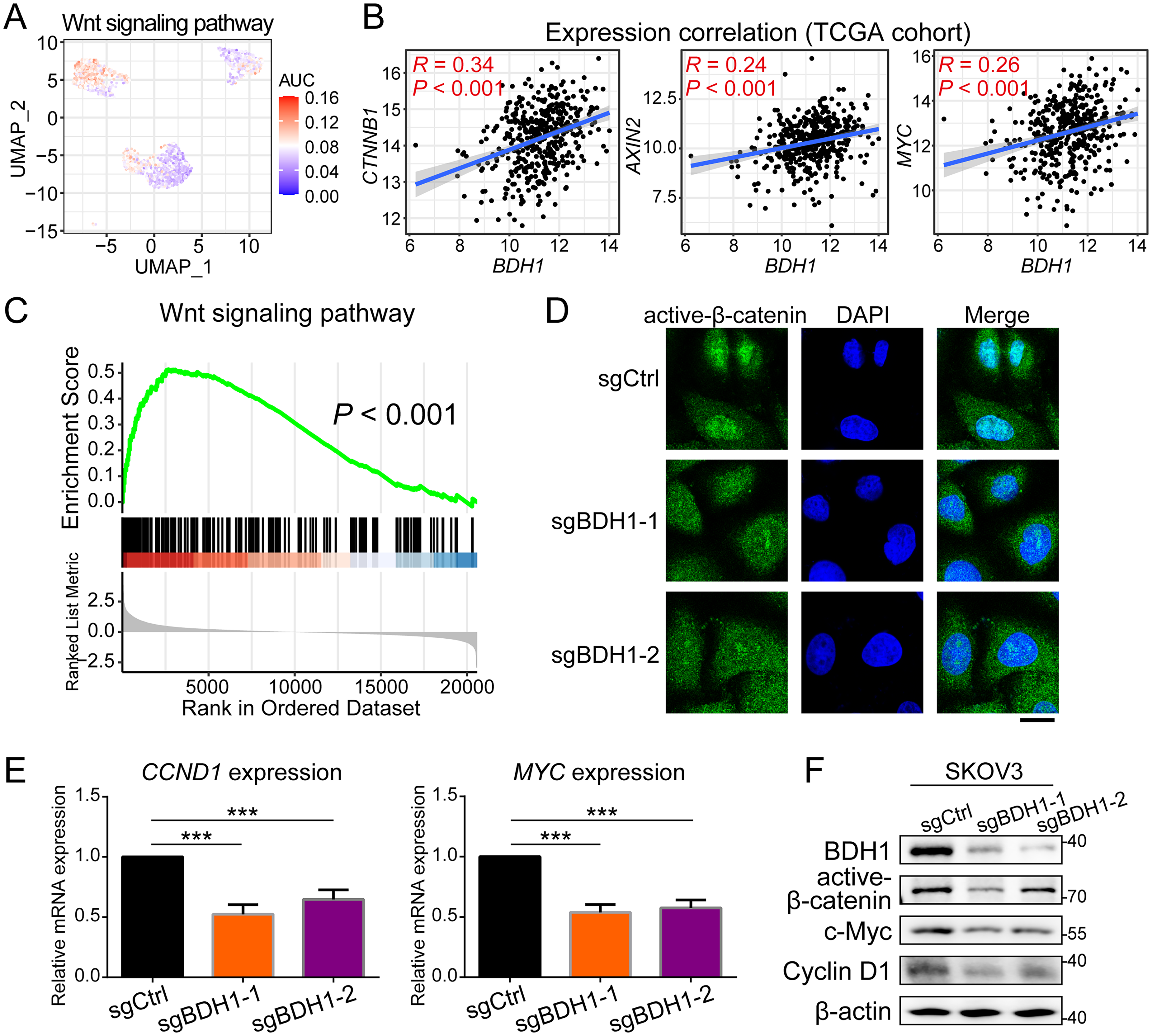
BDH1 promotes the malignance features of OV cancer cells by regulating Wnt signaling pathway. (a) GSVA data demonstrated upregulated Wnt signaling activity in BDH1 + cancer cells. (b) Spearman's correlation revealed positive correlation between BDH1 and Wnt signaling regulators. (c) GSEA results showed that Wnt signaling pathway was enriched in BDH1+ OV samples. (d) The expression of nuclear active-β-catenin was decreased after knocking down of BDH1. (e) The mRNA expression level of CCND1 and MYC was downregulated by BDH1-depletion. (f) The protein expression levels of active-β-catenin, c-Myc, and cyclin D1 were both impaired by BDH1-depletion.
Identification of centrinone as a potential BDH1 inhibitor through virtual screening
A total of 4485 small molecules were evaluated by the virtual screening procedure, and the centrinone was with prime binding affinity with the active site of BDH1 (Supplemental Figure S1(a)). The binding interaction analysis revealed multiple intercellular interactions between centrinone and the active site of BDH1, including conventional hydrogen bond, carbon hydrogen bond, Pi-lone pair, Pi-alkyl bond, and Pi-sulfur bond (Supplemental Figure S1(b)). The MTT assay demonstrated a significant inhibition of centrinone on SKOV3 cell viability with an IC50 of 236.7 nM (Supplemental Figure S1(c)). To add up with, the centrinone treatment significantly decreased the expression level of BDH1 as well as the cell cycling regulators pRb and CDK4 in a dose-dependent manner (Supplemental Figure S1(d)).
Discussion
The β-hydroxybutyrate dehydrogenase 1 enzyme, traditionally recognized for its canonical role in ketone body metabolism through the reversible conversion of β-hydroxybutyrate (β-HB) and acetoacetate, has emerged as a critical player in cancer biology.21,22 While its metabolic functions in maintaining energy homeostasis during starvation and stress conditions are well-established in metabolic disorders like diabetes and neurodegenerative diseases,23,24 our investigation of BDH1's oncogenic roles has expanded significantly. This study provides comprehensive evidence that BDH1 operates at the nexus of metabolic reprogramming, cell cycle regulation, and stemness maintenance in OV progression, making it a compelling therapeutic target for this aggressive malignancy.
Our genomic analysis of the TCGA-OV cohort revealed BDH1 gene amplification in 45 tumor samples, with corresponding mRNA upregulation in 21 cases. This amplification pattern resembles other established oncogenic drivers in OV such as CCNE1 and MYC, known to confer aggressive tumor phenotypes and therapeutic resistance. 25 Notably, BDH1 exhibits a low mutation burden in OV, distinguishing it from other oncogenes frequently altered by point mutations. This pattern reinforces the significance of copy number gain and transcriptional upregulation as key drivers of its pro-tumorigenic functions, positioning BDH1 within a class of amplification-driven cancer dependencies. The clinical significance of BDH1 amplification is underscored by its strong association with advanced disease stages and poorer patient prognosis, suggesting it may serve as both a prognostic marker and therapeutic target. Mechanistically, we found that BDH1 occupies a unique position at the intersection of metabolic and proliferative pathways, and thus coordinating the regulation of key cell cycle proteins, such as pRb, CDK4, and p21, while simultaneously influencing cellular stemness properties. Pseudotime analysis revealed the peak expression of BDH1 during early cell cycle phases, suggesting it functions as a metabolic gatekeeper for cell cycle entry, supported by its strong correlation with proliferation markers, such as CDK1 and CDC45. This dual functionality likely explains the positive selection of BDH1 amplification during tumor evolution, as it simultaneously addresses two fundamental cancer cell requirements: unlimited replicative potential through cell cycle control and metabolic flexibility to survive in challenging microenvironments.
The role of BDH1 in maintaining CSCs represents a particularly significant finding, given the central role of CSCs in therapeutic resistance and disease recurrence. Our multi-platform analysis demonstrated that BDH1 + cells exhibit lower differentiation scores and enrichment of stem-cell-related processes. At the molecular level, BDH1 depletion reduced the expression of key stemness markers, namely CD44, Nanog, SOX2, and KLF4. These findings established a functional link between this metabolic enzyme and the CSC phenotype. Notably, we identified a novel connection between BDH1 and the Wnt/β-catenin pathway, where BDH1 knockdown decreased nuclear β-catenin accumulation. This suggests BDH1 may modulate Wnt signaling activity, potentially through its effects on cellular redox state or acetyl-CoA availability, creating a feed-forward loop that maintains stemness through integrated metabolic and signaling mechanisms. This mechanistic insight helps explain BDH1's particularly strong prognostic value in late-stage patients and suggests that targeting BDH1 could simultaneously deplete the CSC pool while sensitizing tumors to conventional therapies.
The clinical implications of investigating amplified genes like BDH1 are two-fold. First, they may serve as prognostic markers, as demonstrated by our survival analysis showing that the clinical outcomes of BDH1-high patients were significantly worse. This parallels the utility of other amplified genes in risk stratification, such as ERBB2 in breast cancer. 26 Second, amplified genes often encode druggable targets, offering therapeutic opportunities. For instance, PARP inhibitors exploit homologous recombination deficiencies in OV, 27 while AKT inhibitors target the PI3 K/AKT pathway frequently dysregulated in OV patients. 28 Our data position BDH1 as a candidate for similar targeted strategies, especially given its metabolic and signaling roles. Our drug discovery efforts identified centrinone as a promising BDH1-targeting candidate through virtual screening of 4485 small molecules. Although originally developed as a PLK4 inhibitor, 29 centrinone demonstrated prime binding affinity for BDH1's active site through multiple interaction types (hydrogen bonds, Pi-alkyl bonds). Functional validation showed centrinone treatment reduced BDH1 expression, downregulated downstream effectors, and inhibited cell viability. This exemplifies the growing importance of polypharmacology in cancer drug development, where multi-target compounds may offer superior efficacy.30,31 The dual targeting of metabolic regulator BDH1 and mitotic regulator PLK4 could be particularly advantageous in OV, potentially addressing both proliferating bulk tumor cells and quiescent CSCs. Our pathway analysis suggests additional rational combination strategies, as BDH1 + tumors showed activation of PI3K-AKT, TGF-β, and Ras signaling pathways for which targeted inhibitors are in clinical development.
The functional studies were conducted in the SKOV3 cell line, which exhibits endogenous BDH1 expression but does not harbor the BDH1 gene amplification identified in a clinical subset. The observed dependency of SKOV3 cells on BDH1 for proliferation, clonogenicity, and stemness maintenance, as revealed by knockdown experiments, underscores a fundamental oncogenic role for BDH1 that is applicable beyond the context of genetic amplification. This suggests that BDH1 inhibition could have a broader therapeutic window. Nonetheless, the quantitative impact of targeting BDH1 may be most pronounced in tumors with BDH1 amplification, where its expression is highest and likely constitutes a stronger oncogenic dependency. Future studies utilizing patient-derived models or engineered cell lines with BDH1 amplification will be valuable to precisely model the therapeutic vulnerability specific to this genomic subgroup.
Several critical questions also emerge from this work and warrant further investigation. First, the precise molecular mechanisms linking BDH1's metabolic activity to its effects on stemness and cell cycle regulation require further elucidation, particularly whether β-HB production directly influences epigenetic regulation of stemness genes or how BDH1 interfaces with hypoxic microenvironments. Second, while centrinone showed promise, developing more specific BDH1 inhibitors will be essential for fully evaluating the therapeutic potential of BDH1. Third, clinical translation requires validation in patient-derived models and ultimately clinical trials to determine if BDH1 amplification predicts therapeutic sensitivity. Last, the broader implications of targeting amplified metabolic genes in cancer deserve exploration as a potential new paradigm beyond current signaling pathway-focused approaches.
In summary, our study established BDH1 as a multifaceted driver in OV pathogenesis through integrated roles in metabolic reprogramming, stemness maintenance, and microenvironmental interactions. The convergence of these diverse functions in a single target is both biologically unusual and clinically significant, offering opportunities for prognostic stratification and novel therapeutic development. While challenges remain in clinical translation, our integrated genomic, functional, and pharmacological approach provides a roadmap for evaluating other amplified genes in OV and other cancers. As we deepen our understanding of the metabolic foundations of cancer stemness and drug resistance, molecular targets like BDH1 that occupy critical network nodes may prove particularly valuable in overcoming therapeutic resistance in this devastating disease.
Conclusion
This work provides comprehensive evidence establishing BDH1 as a critical oncogenic driver in OV through multifaceted mechanisms. Genomic analysis revealed frequent BDH1 amplification and its significant correlation with poor prognosis. Functional characterization demonstrated that BDH1 promotes tumor progression by regulating cell cycle progression and maintaining cancer stem cell properties. Mechanistically, the oncogenic function of BDH1 is actualized by activating the oncogenic Wnt/β-catenin signaling pathway. Notably, we identified centrinone as a promising therapeutic candidate through virtual screening. Our findings not only advance the molecular understanding of OV pathogenesis but also provide a framework for developing BDH1-targeted therapies, particularly for late-stage patients with poor prognosis.
Supplemental Material
sj-tif-1-jbm-10.1177_03936155261435599 - Supplemental material for BDH1 drives ovarian cancer progression by regulating cell cycling and stemness maintenance through Wnt/β-catenin signaling
Supplemental material, sj-tif-1-jbm-10.1177_03936155261435599 for BDH1 drives ovarian cancer progression by regulating cell cycling and stemness maintenance through Wnt/β-catenin signaling by Shujing Sun, Lianghui Guo and Huili Zhu in The International Journal of Biological Markers
Supplemental Material
sj-tif-2-jbm-10.1177_03936155261435599 - Supplemental material for BDH1 drives ovarian cancer progression by regulating cell cycling and stemness maintenance through Wnt/β-catenin signaling
Supplemental material, sj-tif-2-jbm-10.1177_03936155261435599 for BDH1 drives ovarian cancer progression by regulating cell cycling and stemness maintenance through Wnt/β-catenin signaling by Shujing Sun, Lianghui Guo and Huili Zhu in The International Journal of Biological Markers
Supplemental Material
sj-docx-3-jbm-10.1177_03936155261435599 - Supplemental material for BDH1 drives ovarian cancer progression by regulating cell cycling and stemness maintenance through Wnt/β-catenin signaling
Supplemental material, sj-docx-3-jbm-10.1177_03936155261435599 for BDH1 drives ovarian cancer progression by regulating cell cycling and stemness maintenance through Wnt/β-catenin signaling by Shujing Sun, Lianghui Guo and Huili Zhu in The International Journal of Biological Markers
Footnotes
Ethical considerations
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contributions
Conceptualization, Huili Zhu; Data curation, Lianghui Guo; Formal analysis, Lianghui Guo; Methodology, Shujing Sun; Project administration, Huili Zhu; Resources, Huili Zhu; Supervision, Huili Zhu; Validation, Shujing Sun; Writing—original draft, Huili Zhu.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Supplemental material
Supplemental material for this article is available online.