
Abstract
Introduction
Cervical cancer represents a significant global health problem, ranking as the fourth most prevalent malignant cancer in women, particularly in low- and middle-income countries. Epigenetic silencing via aberrant methylation of tumor suppressor genes’ promoters represent a second hit of cancer initializing, as well as progressing. The aim of current meta-analysis was to systematically evaluate the potential of DNA methylation-based biomarkers in non-invasive or minimal invasive samples using molecular-based approaches for cervical cancer screening and diagnosis.
Methods
The Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guideline was applied to perform our meta-analysis. The frequency, odds ratios, sensitivity as well as specificity with the corresponding 95% confidence intervals were used to assess the effect sizes.
Results
There were 20 eligible articles ultimately included in the current meta-analysis. In this meta-analysis, multiple tumor suppressor genes, such as, especially, PAX1, SOX1, CDO1, GHSR, were shown to undergo hypermethylation in cervical cancer samples compared with controls. Urine samples, when combined with MSP- and qMSP-based approaches, emerged as a particularly effective non-invasive strategy.
Conclusion
The current meta-analysis highlighted the important steps toward establishing DNA-methylation-based biomarkers as accessible and reliable tools for CC screening and diagnosis.
Introduction
Globally, cervical cancer (CC) remains the fourth most common cancer in women, representing a major burden on society, public health, and the economy. According to the World health Organization, there were approximately 660,000 new cases and 350,000 deaths in 2022. 1 The demographic projections indicate a significant rise, with an estimated 35 million new cases of cancer per year anticipated by 2050. 2 Cervical cancer is becoming an increasingly disproportionate burden falling on low- and middle-income countries. More than 80% of CC cases occur in low- and middle-income countries (LMICs), where the age-standardized CC death rate is at least six times higher than in high-income countries. 3 The highest rates of the CC incidence and mortality are in sub-Saharan Africa, Central America, and South-East Asia. 1 This disparity may be partially explained by the multiple systemic issues, such as limited access to high-quality medical care, lack of effective preventive strategies, a lack of trained personnel, etc., faced by LMICs. These data underscore the urgent need for a comprehensive, equity-driven global approach to CC control—one that prioritizes prevention, early detection, and access to treatment, particularly in underserved regions.
In parallel with addressing the inequalities of CC, there is growing interest in elucidating the etiological underpinnings of cervical carcinogenesis, which could be served as novel insights into screening, prognosis, diagnosis of CC, as well as informed the development of potential therapeutic targets. Persistent infection of high-risk human papillomavirus (HPV), which is an epitheliotropic virus belonging to the family of Papillomaviridae, has been identified as the principal causative factor in the cervical carcinogenesis. In particular, HPV-16, HPV-18 are responsible for the majority of CC. 4 It is well documented that the infection of HPV-16, and HPV-18 account for approximately 70% of all incidences of invasive CC worldwide. 5 The overexpression of oncogenic proteins of E6, and E7, which inactivate key tumor suppressors—such as p53 and retinoblastoma protein—through the degradation of E2 are the most largely studied HPV proteins serving as the main drivers of HPV-related cervical carcinogenesis. 6 Although the persistent high-risk HPV infection is a critical initial factor in the development of CC, it is not sufficient on its own to drive the progression to invasive disease. Emerging experimental evidence highlights the significant role of the epigenetic dysregulation, particularly aberrant DNA methylation—the addition of methyl groups onto cytosine residues in a CpG (Cytosine–Guanine) dinucleotide, in the carcinogenesis of CC.7,8 The well-characterized DNA methylation contributing to cervical carcinogenesis is methylated-promoter regions of tumor suppressor genes, which results in their transcriptional silencing and loss of tumor-suppressive functions. 9 In CC, numerous tumor suppressor genes, such as DAPK, SOX1, GSTP1, CDKN2A, PAX1, etc., have been found to undergo the event of aberrant hypermethylation, resulting in the repression of its role of regulating cell cycle arrest, apoptosis, DNA repair, metabolism pathways, and genomic stability.7,10–12 DNA methylation analysis offers several distinct advantages compared to protein-based markers. Methylated DNA may be highly stable and can be detected using polymerase chain reaction (PCR) and PCR-based methods, allowing for the sensitive and specific identification of methylation patterns in bodily fluids, such as blood, urine, or cervical secretions.13–16 Moreover, the phenomena of DNA methylation occurs early and throughout in the carcinogenesis process.15,17 Thus, these data highlight DNA methylation particularly well-suited for non-invasive approaches for cancer early detection, screening, and diagnosis.
An extensive amount of work has demonstrated that numerous methylated genes can serve as effective prognostic or diagnostic biomarkers based on analysis of non-invasive samples found in bodily fluids such as plasma, serum, sputum, urine, or peritoneal fluid.7,10,11,13,15–17 Although the promising results reported in individual studies, DNA methylation-based non-invasive tests have yet to make a full transition from research to routine clinical practice. Recent reports in the literature expose the heterogeneity, due to the sensitives, and intra-/inter-assay coefficients of various populations, methodological strategies, and sources of samples. Therefore, in this systematic review and meta-analysis, we aimed to evaluate the performance of DNA methylation-based biomarkers in non-invasive clinical samples, with the goal of assessing their potential applicability in routine cancer detection and comparing their effectiveness to conventional diagnostic methods.
Materials and methods
Search strategy and selection process
The comprehensive and systematic search were carried out following the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidelines. The data were extracted and collected from PubMed (NCBI), and other databases were searched for publications until April 2025. A combination of Medical Subject Headings (MeSH) terms and free-text keywords were applied in the search strategy and selection process (Supplementary Table S1). Additional studies were retrieved via the lists of references in articles.
Inclusion and exclusion criteria
Since the study aimed to evaluate the performance of DNA methylation-based biomarkers for early diagnosis, screening of CC, the following specific inclusion and exclusion were applied to identify eligible studies for meta-analysis. Studies were included if they met following criteria: (a) The articles were limited to studies written in English; (b) they must have case-control study or cohort design; (c) the studies involve patients who underwent non-invasive screening procedures such as Pap smears, self-collected vaginal samples, or liquid-based cytology; and (d) they must have evaluated one or more DNA methylation biomarkers for cervical screening, or diagnosis, and provided sufficient data for extraction or calculation of sensitivity, specificity, area under the curve (AUC), positive predictive value (PPV), or negative predictive value (NPV).
Studies were excluded if: (a) those articles were written in other languages; (b) they did not evaluate any DNA methylation biomarkers, or used invasive sampling methods such as tissue, biopsies, etc.; (c) articles were review, meta-analysis, letter to editors, conference abstracts, or studies without original data; (d) studies that lacked vital information for analysis; and (e) duplicate or overlapping studies.
Data extraction
To ensure the consistency, transparency, and accuracy in data collection, relevant data and information from each eligible study were systematically extracted by two authors using a standardized data extraction form. This form included predefined fields, including first author's name, year of publication, and geographical region, methodological, clinical parameters, sources of biological specimens, status of DNA methylation, and the techniques or platforms used for detecting methylation. Any discrepancies were solved by discussion or consultation with a third author.
Statistical analysis
An online statistical tool (https://metaanalysisonline.com/) was applied to statistically calculate the candidate genes’ methylation of the case group and control group. 30 The value of odds ratio (OR) and 95% confidence interval (CI) were calculated to evaluate the association between each candidate gene's methylation and cancer risk. Variants in year, ethnicity, population origin, source of samples, and methodological approaches used for methylation detection, were considered potential sources of heterogeneity among the included studies. We used a Cocharn's Q test and I2 statistics to access the heterogeneity. 8 The degree of heterogeneity was assessed with the following interpretation: a value of I2 below 25% indicated no heterogeneity, value between 25% and 50% indicated moderate heterogeneity, and values exceeding 50% reflected substantial heterogeneity. 31 In the case of significant heterogeneity, evidenced by P < 0.05 for the Q test and I2 > 25%, a random-effects model was used. In contrast, if heterogeneity was minimal (I2 < 25%), a fixed-effects model was employed to calculate the pooled odds ratios. Also, a funnel plot was used to test the bias of publications by performing the Begg and Egger's test. 32 All P-values calculations were two-sided, and when P-value < 0.05, it was statistically significant.
Results
Study characteristics: extracted data from the included studies
Our search initially retrieved a total of 945 records through database searches using the predefined keywords (Supplementary Table S1). After screening titles and abstracts, as well as removing duplicates, reviews, meta-analysis, and articles that did not meet the inclusion criteria, 20 eligible articles were ultimately included in the current meta-analysis. (Supplementary Table S2). Our study showed that studies had been performed in Asia (12 articles) including China (4), Vietnam (3), India (2), Korea (1), Taiwan (1), Hong Kong (1); in Europe (7 articles) including Austria (4), the Netherlands (3), Germany (1); and in Africa (Senegal, 1 article). This distribution reflected a strong global effort in exploring DNA methylation-based biomarkers, with a strong concentration of studies originating from Asian and European countries. Current data pointed out the usage of various non-invasive or minimally invasive samples, including serum, plasma, urine, blood, Pap smear, and swab, reflecting ongoing efforts to develop accessible non-invasive screening methods for CC. A wide range of 55 tumor suppressor genes (TSG), including CDH1, CHD13, p14, p15, p16, SST, GHSR, DAPK, RARβ, FHIT, DcR1, etc., were examined as a comprehensive strategy to identify a non-invasive biomarker for CC detection and screening. Additionally, 15 of the 22 studies (accounting for 68.18%) investigated multiple candidate genes, defined as more than two targets, for DNA methylation analysis to enhance the sensitivity and specificity of CC detection and screening. Among them, the study of Jha et al. 33 explored 13 candidate genes, including p14, p15, p16, p21, p27, p57, p53, p73, RARβ2, FHIT, DAPK, STAT1, RB1, by using method of methylation-specific PCR (MSP) to evaluate the methylation status of their promoter methylation. These comprehensive multiple gene investigations aimed to uncover reliable methylation-based biomarkers for CC detection by focusing on genes involved in cell cycle regulation, apoptosis, and tumor suppression.
Meta-analysis: methylation profile in candidate genes for CC
An overview of the key findings from the enrolled case-control studies included in this review comprehensively outlined in Supplementary Table S2. Twelve appropriate studies investigated 34 candidate genes (SOX1, PAX1, LMX1A, NKX6-1, WT1, ONECUT1, hTERT, TIMP3, GHSR, SST, ZIC1, DAPK, MGMT, CADM1, MAL, p16, CDH1, MEG3, CDH13, SOCS1, GSTP1, HSPA2, MLH1, RASSF1A, SOCS2, SIM1, RARβ, FHIT, TWIST1, ADCYAP1, BHLHE22, CDO1, GALR1, and HAND2). A meta-analysis was conducted for each candidate gene, which was investigated in at least two independent studies or across two different sample types within a single article. A quantitative meta-analysis, then, was performed to estimate the pooled methylation frequencies in both cancer and non-cancer samples. Studies included in the meta-analysis are marked with an asterisk (*). An overview of the key findings from enrolled the case-control studies included in this current review was shown in Supplementary Table 2. The L’Abbé plot, shown in Figure 1, provides a comparative overview of the methylation percentage observed across 14 candidate genes in CCs compared to non-tumor control samples. As presented, the diagonal line represents the point of equivalence, where the methylation percentage is equal in CC and non-tumor samples. Notably, all 14 candidate genes are positioned above this line, signifying consistently higher methylation levels in CC samples. This pattern demonstrates the tendency of these genes to become hypermethylated in CC, supporting their significant role as epigenetic biomarkers for CC screening, and diagnosis.
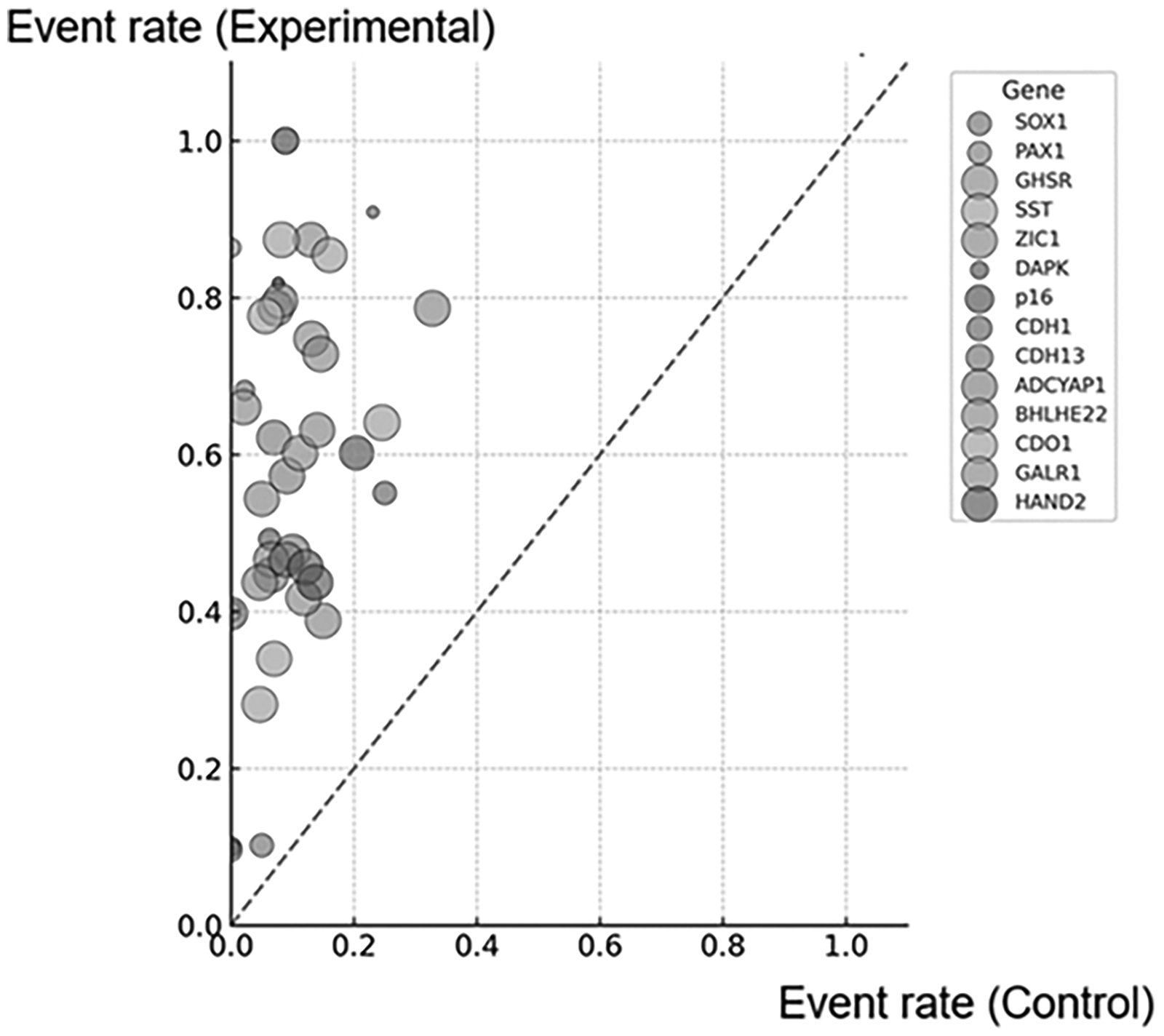
L’Abbé plot for the event rate of control (non-tumor) and experimental (tumor). Each symbol represents one gene within one study with the size of the symbol corresponding to the weight of the study. The diagonal line indicates where the methylation percentage is equal in the cervical sample and non-cancerous samples.
Figure 2 represents the pooled methylation frequencies in non-invasive/minimally invasive cancerous samples and non-cancerous samples across the included studies. For most candidate genes, the pooled estimates reveal markedly higher methylation frequencies in cancerous samples than in controls, underscoring the potential role of these epigenetic alterations as cancer-specific biomarkers. In tumor-bearing samples, the random-effect model was applied to estimate the pooled methylation frequencies of SOX1, GHSR, SST, ZIC1, DAPK, P16, CDH1, CDH13, ADCYAP1, BHLHE22, CDO1, and GALR1, due to the moderate to high heterogeneity (I2 > 25%). In contrast, the fixed-effect model was applied for genes, including PAX1, and HAND2, with low or no heterogeneity, yielding more precise estimates to yield more precise estimates. As presented in Figure 2, the highest pooled methylation frequencies in cancerous samples were observed in PAX1 (0.92, 95% CI: 0.74–1.00), CDO1 (0.84; 95% CI: 0.78–0.89), GHSR (0.81; 95% CI: 0.73–0.87), and SOX1 (0.79; 95% CI: 0.47–1.00), while the lowest was recorded for p16 (0.33; 95% CI: 0.00–0.84). For control samples, the pooled methylation frequencies of SOX1, PAX1, SST, ZIC1, P16, CDH1, BHLHE22, CDO1, DAPK, and GALR1 were estimated using the random-effects model due to moderate to high heterogeneity. Conversely, the fixed-effect model was used for GHSR, CDH13, ADCYAP1, and HAND2, where heterogeneity was low or absent. Control samples exhibited low methylation frequencies across all candidate genes, ranging from 0.02 for DAPK (95% CI: 0.00–0.11) to 0.16 for ZIC1 (95% CI: 0.04–0.34). Additionally, the significant difference between case and control for genes including PAX1, SOX1, GHSR, and CDO1 were observed, reinforcing their potential for cancer diagnosis and screening.
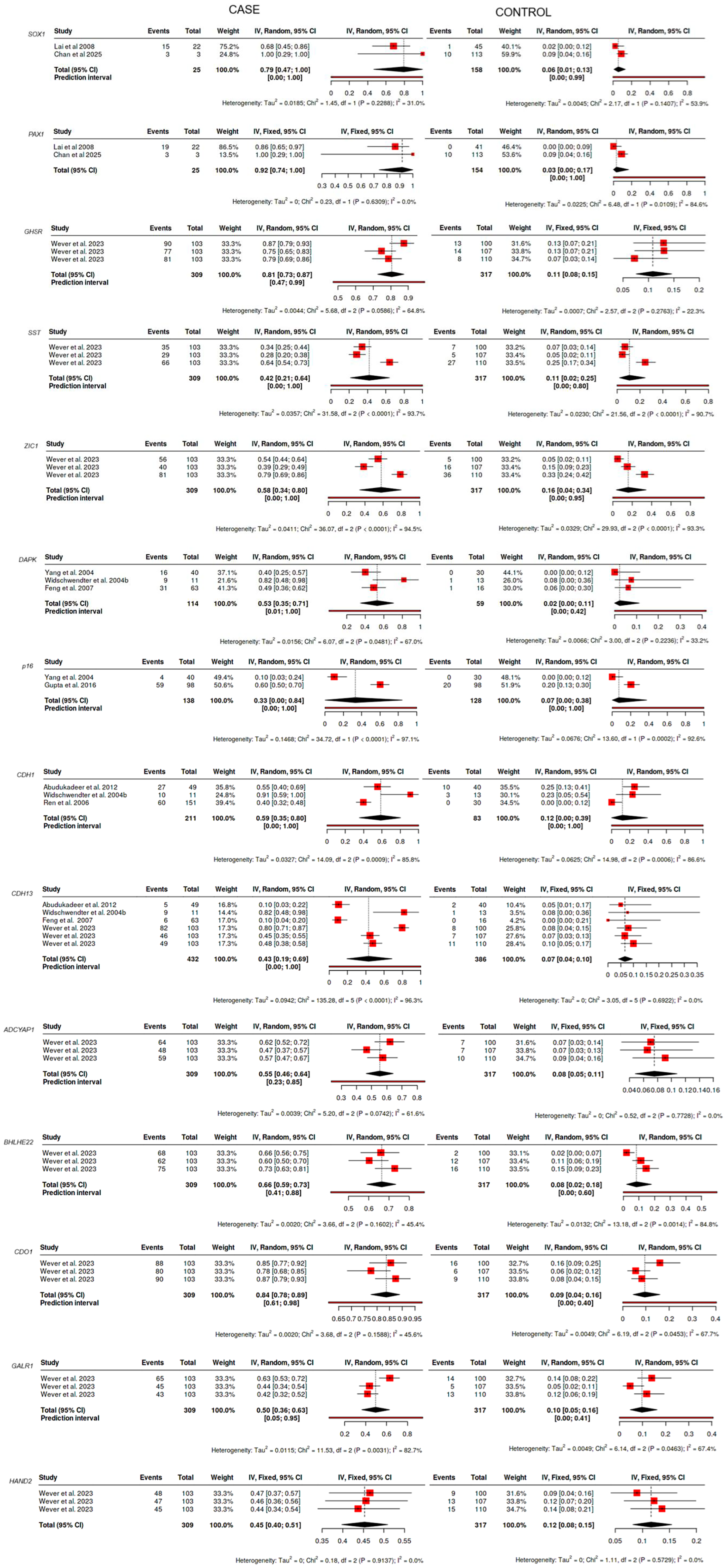
Forest plots of candidate genes’ methylation frequency in cancerous samples (Case) and non-cancerous samples (control).
The pooled OR analysis of methylation profiles in candidate genes for CC
The pooled OR for with 95% CI for 14 candidate genes was presented in Figure 3. The pooled OR (OR > 1.0) results exhibited markedly elevated associations with gene methylation and CC. The fix effect model was applied to evaluate the OR of genes, including SOX1, PAX1, SST, DAPK, p16, ADCYAP1, CDO1, and HAND2, with no heterogeneity, or low heterogeneity (I2 < 25%). In contrast, the random effects model was used for genes GHSR, BHLHE22, CDH1, CDH13, GALR1, and ZIC1 due to the high heterogeneity. Among them, the highest pooled ORs were observed for PAX1, and SOX1, counting for OR = 217.21, 95% CI: 27.74–1703.12, and OR = 119.78, 95% CI: 14.25–1006.73, respectively. The consistently highest pooled ORs, combined with their absence of heterogeneity, for PAX1 and SOX1, suggesting these genes may serve as particularly strong epigenetic biomarkers for CC detection.
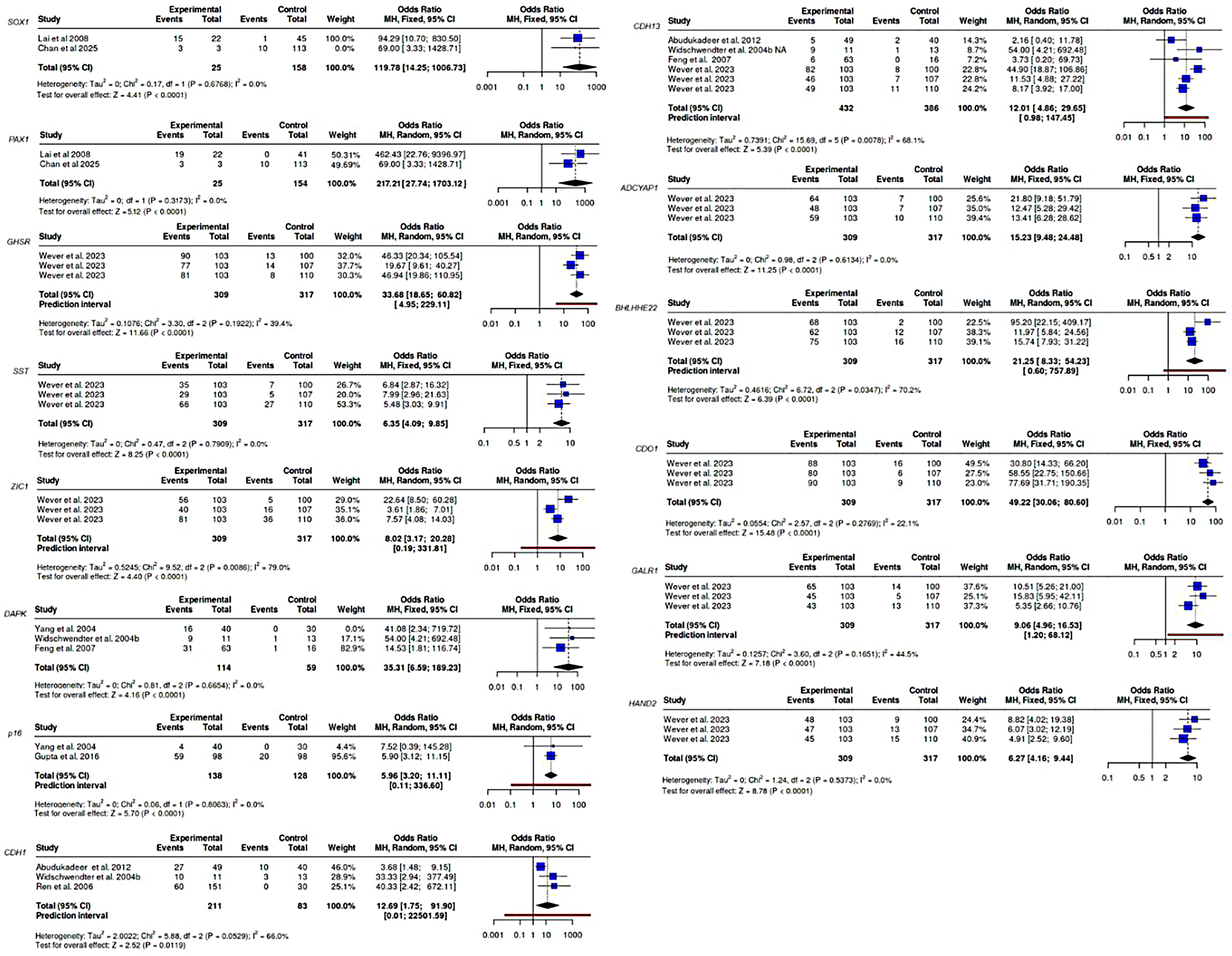
The pooled OR with corresponding 95% CI for each candidate gene is found under the random effect model or fixed effect model.
Subgroup analysis: sources and methylation detection methods in CC screening and diagnosis
Across the included studies, the sample types spanned a spectrum: from non-invasive, including urine, Pap smear, swab, scrape, and cervicovaginal self-samples to minimally invasive samples, including blood-derived specimens, such as plasma, and serum. As indicated in Figure 4, and Table 1, the non-invasive specimens, especially urine, are the predominant sources for DNA methylation analysis of 10 target genes associated with CC. Regarding methylation detection methods, MSP-based approaches were the most prevalent, accounting for 66.66% overall. Of these, standard MSP made up the majority at 44.44%, while qMSP contributed an additional 22.22%. Methylight and DHPLC-PCR accounted for 22.22%, and 11.11%, respectively. Additionally, the integration of qMSP or MSP with urine specimens yielded the one of the most effective screening approaches for evaluating the methylation status of candidate genes.
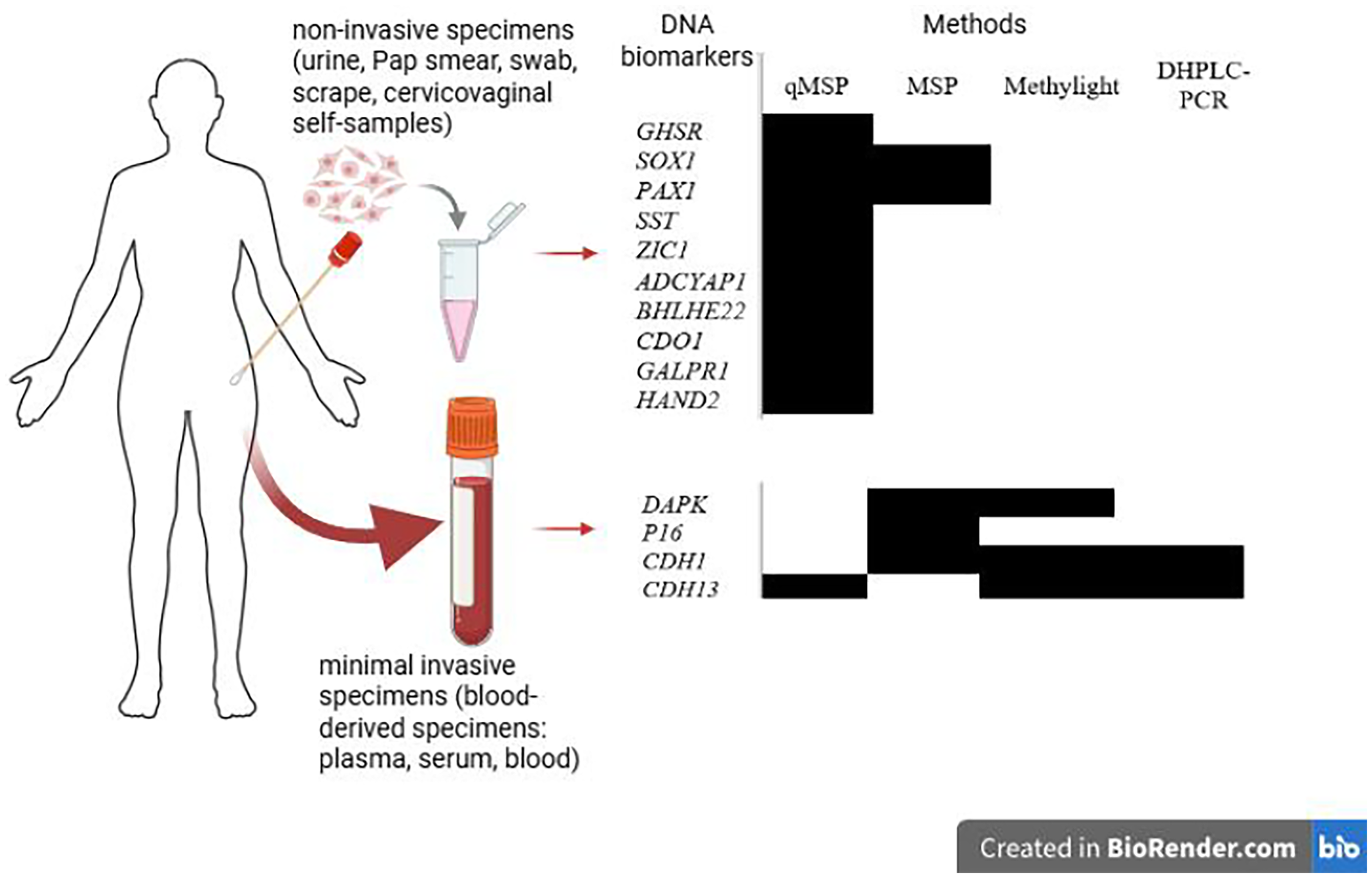
DNA methylation biomarkers by sample sources, assay methods, and target genes. The black square represented the method used to detect methylation of the target gene.
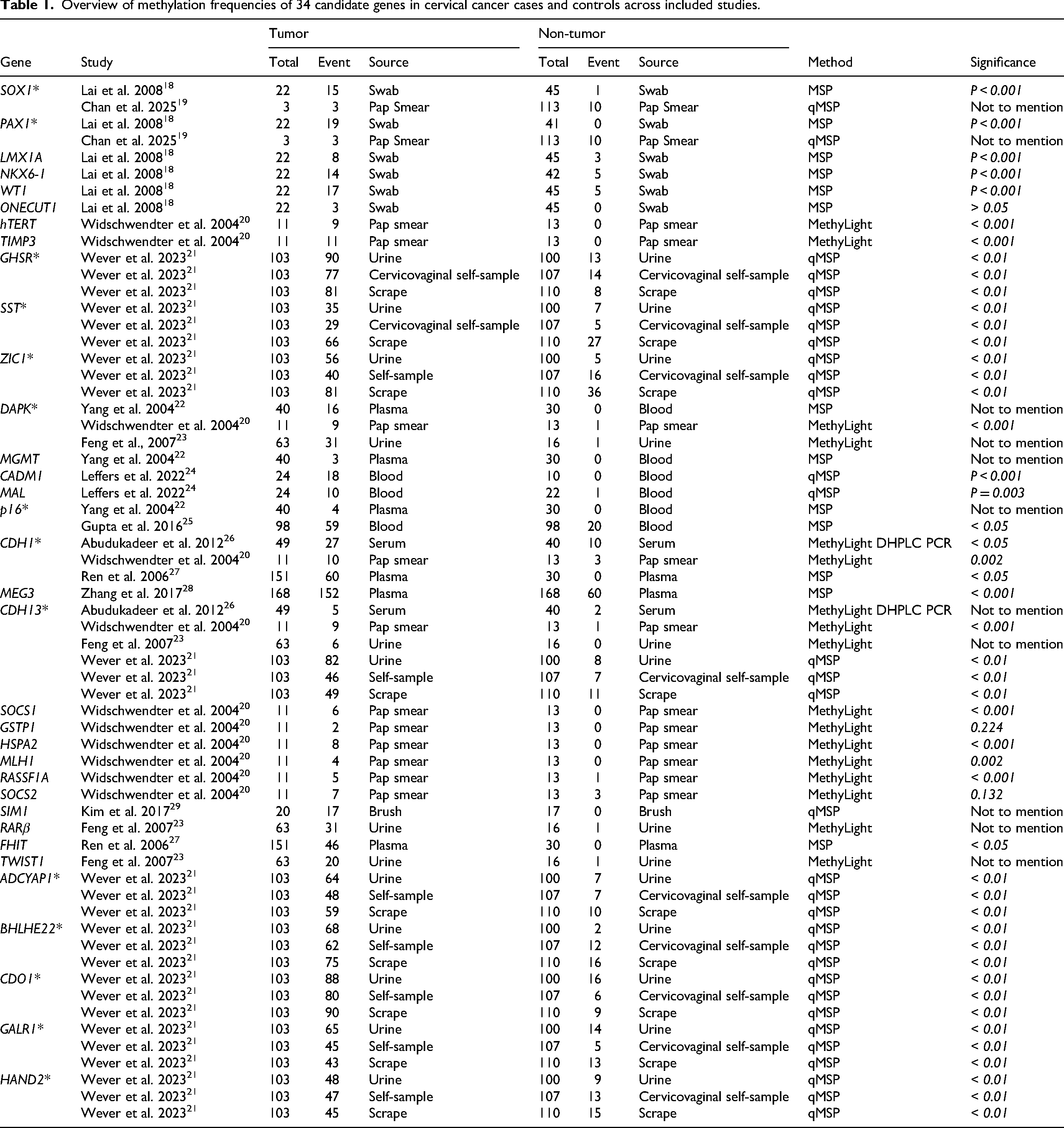
Overview of methylation frequencies of 34 candidate genes in cervical cancer cases and controls across included studies.
Functional classification of methylated genes
The functions of 14 included candidate genes—most of which act as tumor suppressor genes involved in diverse biological pathways—including regulating cellular proliferation, apoptosis, differentiation, and invasion. Several genes, such as SOX1, CDH1, and CDH13, modulate the Wnt/β-catenin signaling pathway, influencing cell adhesion, polarity, and growth suppression. Others, including PAX1 and ZIC1, inhibit proliferative kinase cascades and modulate the MAPK pathway, while GHSR—not classified as a TSG—has been linked to tumor severity via the PI3 K/Akt and GHRH-GH-IGF pathways. Genes like SST, DAPK, and p16 regulate apoptosis and cell cycle control through pathways such as Notch1, cell death signaling, and CDK6-HuR-IL1A. ADCYAP1 and BHLHE22 are associated with PACAP and immune-related pathways, respectively, contributing to cell fate determination and immune regulation. Additionally, CDO1 promotes ferroptosis, and GALR1 suppresses colony formation, though their pathways remain unclear. Notably, HAND2 delays CC progression through VEGFA signaling. Collectively, these methylated genes underscore the complex, multi-pathway suppression of oncogenesis in CC.
Discussion
Recently, research on the phenomenon of DNA methylation (an epigenetic process where a methyl group (-CH3) is added to a DNA molecule) in CC has grown substantially, reflecting its importance in development of diagnosis and prognosis of CC. It can be explained that the methylation of tumor suppressor genes are strongly associated with cervical tumor initiation, progression, and metastasis.9,11,26,34–37 The current meta-analysis incorporated 20 eligible studies published up to the year 2025, underscoring an updated and comprehensive evaluation of methylation-based biomarkers, with a particular focus on those utilizing non-invasive or minimally invasive sample types for CC diagnosis and prognosis. Therefore, these results of meta-analysis establish a strong scientific and fundamental foundation for its application in clinical practice, and development of an accurate, non-invasive diagnostic approach aimed at facilitating early detection, improving prognostic assessment, and ultimately enhancing patient outcomes in CC management.
The initial finding of this review highlighted the compelling evidence for consistent hypermethylation patterns across 14 candidate genes, significantly demonstrating that higher methylation frequencies in CC compared to non-tumor controls, as illustrated in the L’Abbé plot (Figure 1). It suggested their potential as pivotal epigenetic biomarkers in CC development and diagnosis. The fixed-effects model and random-effects model were applied to evaluate the pooled methylation and ORs aiming to appropriate interpretation of the statistical associations across studies. 38 As presented in Figure 2, the pooled methylation frequency analysis demonstrated that a strong distinction between cancerous and non-cancerous samples across the candidate genes, reinforcing the role of aberrant DNA methylation, considering as a cancer-specific hallmark. 9 The random-effects model was applied to genes including SOX1, GHSR, SST, ZIC1, DAPK, p16, CDH1, CDH13, ADCYAP1, BHLHE22, CDO1, and GALR1 reflecting the moderate to high heterogeneity observed across included studies as a result of the demographics, sample types, etc. Therefore, it is suggested that there is a need for methodological standardization in future research. In contrast, PAX1 and HAND2 exhibited less heterogeneity, providing more robust and consistent methylation estimates across studies. Among the candidate genes, PAX1 displayed the highest pooled methylation frequency in cancerous samples (0.92; 95% CI: 0.74–1.00), followed closely by CDO1 (0.84; 95% CI: 0.78–0.89), GHSR (0.81; 95% CI: 0.73–0.87), and SOX1 (0.79; 95% CI: 0.47–1.00). These values underscore the strong association between methylation of these genes and cervical carcinogenesis, supporting their candidacy as primary biomarkers for screening and early detection. PAX1 (paired box gene 1), SOX1 (sex-determining region Y-related high-mobility group box 1), CDO1 (cysteine dioxygenase 1), and GHSR (growth hormone secretagogue receptor) have been implicated in many types of cancer progression, as they are frequently silenced through promoter hypermethylation. PAX1, a tumor suppressor gene belongs to the PAX family, has been proposed to take part in the Notch pathway. Its activation is blocked by the silence of the HOX family of transcription factors, which are downstream targets of Notch signaling. SOX1 has been reported to inhibit cervical, hepatic, and nasopharyngeal carcinomas through modulation of the Wnt/β-catenin signaling pathway.39–41 CDO1 gene is a non-heme structured, iron-containing metalloenzyme involved in the conversion of cysteine to cysteine sulfinate, and plays a key role in taurine biosynthesis and induce apoptosis. According to the hypothesis of Brait et al., 42 they prove that cancer-specific methylation and the decrease of CDO1 expression may be common events in human cancer development. Meanwhile, hypermethylation of GHSR has been reported as a common epigenetic alteration of high diagnostic value in a broad spectrum of cancers. 43 Ghrelin is a 28-amino acid peptide which serves as the endogenous ligand of the growth hormone secretagogue receptor (GHSR), has been implicated in cancer progression and metastasis via the ghrelin–GHSR signaling axis, with aberrant methylation contributing to dysregulated pathway activity. 44 Collectively, these biological mechanisms, together with the high methylation frequencies observed in cancerous tissues, provide strong evidence for a robust association between methylation of these genes and cervical carcinogenesis, supporting their candidacy as primary biomarkers for CC screening and early detection. Conversely, p16, a widely studied tumor suppressor gene, showed the lowest pooled methylation frequency (0.33; 95% CI: 0.00–0.84), suggesting that its diagnostic utility as a single methylation marker may be limited, though it could retain value in multi-gene panels.
Notably, our pooled OR values of candidate genes were higher than 1.0, suggesting that individuals with the gene methylation had the higher odds of disease, thereby demonstrating a positive correlation between aberrant methylation and disease presence. 45 Among them, the pooled OR of PAX1 and SOX1 showed the highest values at 217.21, and 119.78, with the significant 95% CI, respectively. It meant that methylation of these genes were strongly associated with a 217.21-fold and 119.78-fold increased likelihood of disease, respectively, within no heterogeneity (I2 = 0.00%, fix effects model), largely due to their involvement in proliferation suppression and Wnt/β-catenin pathway regulation.40,41
From a subgroup analysis perspective, the sources of samples highlighted a notable trend toward the application of non-invasive/minimal invasive specimens for CC screening and diagnosis. Among these sources, urine emerged as the most frequently utilized source for DNA methylation analysis of candidate genes, particularly combined with the methods of qMSP or MSP, offered a sensitive and easily accessible approach for CC screening and diagnosis. According to the study of Snoek et al., 15 CC screening using urine offered advantages over conventional cervical scrapes and self-samples, which relates to the fact that the use of urine is expected to largely increase screening attendance. Despite certain limitations, the MSP-based methods, including MSP and qMSP, remain valuable techniques for detecting and evaluating DNA methylation patterns, due to their simple design, implementation, high sensitivity, and cost-effective capacity. 46 Therefore, the use of urine in combination with MSP or qMSP provided an easy-going approach that could serve as a potential alternative to physician-collected or self-collected cervical samples for evaluating the methylation of candidate. Our findings support the clinical translation of selected DNA methylation biomarkers, especially PAX1, SOX1, CDO1, and GHSR, into non-invasive screening strategies. Future research should prioritize prospective, large-scale, multi-center validation studies across diverse populations. Furthermore, the standardization of assay protocols and the definition of cut-off thresholds will be critical for regulatory approval and successful implementation in population-based screening programs.
Conclusion
To date, several significantly differentially methylated gene patterns have been identified in CC aiming to establish accessible, and reliable biomarkers for CC screening and diagnosis. Thereby, it is important to conduct a meta-analysis to systematically evaluate the potential of these candidate genes across diverse studies and populations. Such an approach not only synthesizes current evidence but also addresses heterogeneity in methodologies, sample sources, and assay platforms. As a result, the signatures of methylation, consistently higher methylation frequencies, and odds ratios were calculated in cancerous samples compared to non-cancerous controls. Particularly, candidates including PAX1, SOX1, CDO1, and GHSR highlighted their potential utility in non-invasive/minimally invasive specimens, such as urine, for establishing the strategies of CC screening and diagnosis. The urine sample in combination with MSP- and qMSP-based approaches demonstrated a high number of uses, emerging as one of the most effective non-invasive strategies for evaluating the methylation status of candidate genes.
Supplemental Material
sj-docx-1-jbm-10.1177_03936155261419529 - Supplemental material for Meta-analytic and systematic review of the diagnostic value of DNA methylation-based biomarkers in cervical cancer
Supplemental material, sj-docx-1-jbm-10.1177_03936155261419529 for Meta-analytic and systematic review of the diagnostic value of DNA methylation-based biomarkers in cervical cancer by Hue Hong Thieu, Thuy Ai Huyen Le and Thuan Duc Lao in The International Journal of Biological Markers
Footnotes
Author contributions
Conceptualization, methodology, validation, formal analysis, investigation, data curation: H.H.T., T.A.H.L., T.D.L; Writing—original draft: T.D.L. Writing—review & editing: T.A.H.L., T.D.L. All authors have read and agreed to the published version of the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding authors.
Supplemental material
Supplemental material for this article is available online.