
Abstract
Background
The mechanisms underlying the occurrence and progression of metabolic dysfunction-associated steatohepatitis (MASH)-related liver fibrosis remains poorly understood. This study aims to identify key transcription factors involved in the development of liver fibrosis in MASH patients, thereby providing potential targets for drug discovery.
Methods
Microarray data were retrieved from liver biopsy specimens of MASH patients exhibiting varying stages of fibrosis via the Gene Expression Omnibus database. Differentially expressed transcription factors (DETFs) were identified through the application of Weighted Gene Co-expression Network Analysis. A set of in vitro and in vivo experiments were conducted to investigate the role of MEOX1 in MASH-related fibrosis. To delineate the potential mechanisms, the transcriptomic RNA sequencing (RNA-seq), Alphafold, and PyMOL were used.
Results
A total of six DETFs (MEOX1, SOX4, LEF1, SOX9, MYC, and CBX2) were identified as being positively correlated with the progression of MASH-related fibrosis. MEOX1 was increased in mouse model of MASH diet-induced liver fibrosis and hepatic stellate cells (HSCs) stimulated by transforming growth factor-β1. Knockdown of the MEOX1 markedly suppressed the activation, proliferation, and migration of HSCs. RNA-Seq analysis identified serine protease inhibitor family E member 1 (SERPINE1) as the critical target of MEOX1 within HSCs. The protein interaction sites of MEOX1 and SERPINE1 were predicted using Alphafold and PyMOL.
Conclusion
In summary, as a pivotal transcription factor, MEOX1 activates HSCs via SERPINE1, thereby promoting liver fibrosis associated with MASH. Inhibition of the MEOX1-SERPINE1 pathway could offer a novel therapeutic avenue for treating MASH-related fibrosis.
Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) has currently emerged as the most prevalent chronic liver disorder globally, affecting approximately 25–38% of the adult population. 1 As a form of metabolic liver injury closely related to insulin resistance and genetics, up to 25% of patients with MASLD will advance to metabolic dysfunction-associated steatohepatitis (MASH), which is currently anticipated to become the second leading cause of liver transplantation.2–4 The hallmark of MASH is hepatic fibrosis. Studies indicate that approximately 75% of MASH patients will progress to liver fibrosis, with 34.5% developing advanced fibrosis (stages fibrosis-3 or 4). This progression markedly elevates the risk of mortality from liver-related and cardiovascular diseases.5–7 Hence, it is of crucial significance to elucidate the mechanisms of fibrosis development and resolution in MASH and to discover effective therapeutics to prevent the progression of MASLD and MASH-related fibrosis.
In susceptible individuals, factors like insulin resistance result in elevated levels of free fatty acids and increased oxidative stress in the liver, subsequently giving rise to mitochondrial dysfunction, endoplasmic reticulum stress, as well as hepatocyte injury and apoptosis. 8 The injured and necrotic hepatocytes release cytokines and damage-associated molecular patterns, which attract and activate immune cells and hepatic stellate cells (HSCs). 9 This activation leads to the continuous production and deposition of collagen and extracellular matrix (ECM), disrupting the normal architecture and function of the liver, thereby facilitating the progression of fibrosis.10,11 In models of diet-induced MASH, in response to chronic liver injury, the activation of HSCs is the core link and key driving factor for the occurrence and development of liver fibrosis. 12 How to reverse the activation of quiescent HSCs and promote the apoptosis of activated HSCs is the main strategy and direction for anti-fibrosis treatment.
Currently, there is a growing focus on the role of transcription factors (TFs) in the development of pharmacological interventions for MASLD/MASH and advanced fibrosis. 13 Several TFs, such as peroxisome proliferator-activated receptor, 14 farnesoid X receptor, 15 forkhead box O1, 16 sterol regulatory element binding protein-1c, 17 and interferon regulatory factor 1 18 mediate the progression of MASLD/MASH by regulating lipid metabolism, insulin resistance, oxidative stress, immune inflammation and so on. Most current studies concentrate on the regulation of lipid metabolism by TFs, while limited research has specifically addressed the inhibition of fibrosis progression associated with MASH. Considering the poor prognosis of MASH-related fibrosis, it is necessary to investigate critical TFs that can inhibit the fibrotic process and facilitate the regression of hepatic fibrosis.
In the present study, we explore the differentially expressed genes (DEGs) and differentially expressed transcription factors (DETFs) associated with MASH-related fibrosis by using bioinformatics analysis. Furthermore, we verify the correlation between DETFs and the extent of MASH-related fibrosis by employing diverse mouse models. Then, our research demonstrates that mesenchyme homeobox 1 (MEOX1) is upregulated in MASH-related fibrosis, and higher expression levels show higher stage of fibrosis, suggesting that it is a potential biomarker and drug target for MASH-related fibrosis progression. Finally, we also reveal that MEOX1 regulates HSCs activation, proliferation, and migration mainly by inducing the expression of serine protease inhibitor family E member 1 (SERPINE1, the gene encoding for plasminogen activator inhibitor-1, PAI-1).
Materials and methods
Data acquisition
We obtained the messenger RNA (mRNA)-expressing profiles of hepatic steatosis, MASH, and hepatic fibrosis from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/). The participants satisfied the following criteria: (a) presence of fatty liver by ultrasonography and other liver imaging test; (b) diagnosed with histologically hepatic steatosis and MASH liver by biopsy; (c) elevated levels of liver enzymes were observed; and (d) interference from excessive alcohol consumption, drug injury, viral hepatitis, autoimmune hepatitis, and other factors were excluded. The liver samples were embedded in paraffin blocks following standard procedures and stained with hematoxylin and eosin (H&E) and Masson's trichrome stains. All liver biopsies were centrally evaluated by an experienced hepatopathologist at one time by using the MASH Clinical Research Network histological scoring system and the hepatic fibrosis was scored on a 5-point scale (0–4) according to the Kleiner classification.19,20 According to the inclusion and exclusion criteria, GSE135251 19 with 53 hepatic steatosis patients and 153 MASH patients (32 stage F0–1 MASH patients, 53 stage F0–1 MASH patients, 54 stage F3 MASH patients, 14 stage F4 MASH patients), and GSE167523 20 with 51 hepatic steatosis patients and 47 MASH patients were enrolled in this study. We conducted a comprehensive analysis of DEGs in liver tissues from patients with hepatic steatosis and MASH across GSE135251 and GSE167523. Furthermore, we investigated the DEGs associated with the stages of MASH-related fibrosis across GSE135251.
For each dataset, to guarantee the integrity and comparability of the raw expression data, we download the data that underwent a standardized preprocessing protocol. Regarding transcriptomic RNA sequencing (RNA-seq) datasets, diverse R packages were employed for the comprehensive subsequent analysis of DEGs.
Weight gene co-expression network analysis
Weight gene co-expression network analysis (WGCNA) was employed to explore the interactions among the genes and to establish potential modules related to MASH-related fibrosis. 21 Specifically, the sample data are initially preprocessed to remove outliers and were utilized to construct a correlation matrix with the assistance of the WGCNA R package. We then selected the optimal soft-thresholding power (β) to transform the correlation matrix into an adjacency matrix. Finally, we employed the hierarchical clustering analysis and the dynamic tree-cutting algorithm to group genes with similar expression patterns into distinct gene modules, and to evaluate the gene significance and module membership. The intersections of genes in the significant module of WGCNA and DEGs were regarded as common genes.
Animals and treatments
A total of 18 eight-week-old male C57BL/6 mice (weighing 20–23 g) were obtained from Shanghai Laboratory Animal Company (SLAC, Shanghai, China). All animal experiments were approved by the Animal Care and follow through the guidelines of the Committee at Shanghai Tongji Hospital (approval no. TJHBLAC-2019-024). The mice were randomly divided into three groups as follows: (a) the model of hepatic steatosis group (n = 6), in which mice were fed with a high-fat diet (HFD) for 16 weeks; (b) the model of MASH group (n = 6), in which mice were fed with methionine-choline-deficient (MCD) diet for 12 weeks; and (c) the chow group (n = 6), in which mice were fed with a standard chow diet for 16 weeks. All mice were housed in a specific-pathogen-free environment with a 12:12-h light/dark cycle and ad libitum access to food and water.
The criteria defined for exclusion of mice during the experiment were as follows: significant reduction in food or water intake, wrinkled fur or self-inflicted harm, waddling, dyspnea, inability to stand, and lack of response to external stimuli. No mice were excluded from the experiment based on the aforementioned criteria.
At the end of the experiment, mice were euthanized with 5% isoflurane for 5 min. The absence of pedal and eyelid reflexes was confirmed to verify unconsciousness. Subsequently, the mice were euthanized through exsanguination under anesthesia to ensure death, which was affirmed by the cessation of respiration, heartbeat, and response to external stimuli. Then, tissue samples for analysis were collected.
H&E staining was used to assess tissue morphology. Collagen deposition was evaluated through Masson's trichrome staining. Serum levels of alanine amino transferase (ALT), aspartate amino transferase (AST), and triglyceride (TG) were measured using enzymatic procedures according to the manufacturers’ instruction (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Cell culture and treatment
Under the condition of 5% CO2 and 37°C, human immortalized hepatic stellate LX-2 cells (Wuhan Pricella Biotechnology Co. Ltd, Wuhan, China) and HepG2 cells (The Cell Bank of Type Culture Collection of The Chinese Academy of Sciences) were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplement with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin solution. LX-2 were maintained in FBS-free DMEM overnight, and then exposed to 10 ng/ml of transforming growth factor-β1 (TGF-β1, PeproTech, Thermo-Fisher Scientific, Waltham, MA, USA) for 24 h to induce activated HSCs. Also, 0.5% bovine serum albumin (BSA) (Sigma-Aldrich, Burlington, MA, USA), free fatty acids (FFA, 1 mM) and palmitic acid (PA, 1 mM) were involved in the starved HepG2 for 24 h to establish an in vitro model of control and steatosis hepatocyte. The small interfering RNAs (siRNAs, RiboBio) were transiently transfected by Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's instructions.
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA samples were extracted from liver tissues, HepG2, and HSCs using Trizol reagent (Sigma-Aldrich, Merck KGaA) and Total RNA Isolation Kit (Vazyme, Nanjing, Jiangsu, China). One microgram of total RNA was reverse transcribed into complementary DNA (cDNA) with qRT-PCR reaction kit (Vazyme) in accordance with the manufacturer's instructions. SYBR Green PCR Master Mix (Vazyme) was employed to synthesize cDNA and QuantStudio 6 Operating Software was utilized to analyze the data. Meanwhile, β-actin in parallel for each run was taken as an internal control, and the mRNA expression levels of target genes were quantified via 2−ΔΔCT method. Each experiment was performed in triplicate. The primers synthesized by Sangon Biotech (Shanghai, China) are listed in Table S1.
Western blot assay
Total proteins were extracted from HepG2, LX-2 cells and mice liver tissues in RIPA (Beyotime) at 4°C and were qualified by BSA assay (Epizyme Biotech, Cambridge, MA, USA). Equal quantities of total proteins were separated by 10% SDS-PAGE and were transferred to polyvinylidene fluoride membrane. Next, the membrane was blocked with 5% nonfat milk dissolved in Tris-Buffered Saline with Tween buffer for 60 min at room temperature. After incubation with the primary antibody (shown in Table S2) at 4 °C overnight, horseradish peroxidase-conjugated secondary antibodies (Beyotime) were used to incubate the membrane for 60 min at room temperature. Finally, the enhanced chemiluminescence (ECL) detection reagent and the ECL system were utilized for the imaging of gels and blots in accordance with the manufacturer's protocol. Each experiment was performed in triplicate.
Oil red O staining
After being treated with PA for 24 h, the medium of HepG2 was removed and then the cells were fixed with 4% paraformaldehyde for 30 min. Next, the diluted oil red O (0.6% oil red O in isopropanol: H2O = 3:2) was added to the HepG2 for 1 h. After being washed with phosphate buffered saline (PBS) three times, an optical microscope was employed to observe the formation of lipid droplets. Each experiment was performed in triplicate.
Cellular immunofluorescence staining
LX-2 cells were plated in a 24-well plate. After being washed with PBS three times, the cells were fixed with cold 4% paraformaldehyde solution at 4°C for 20 min and permeabilized with 0.3% Triton-100 (Beyotime) for 10 min. Subsequently, after being blocked with 1% BSA solution for 30 min, the primary antibody (α-SMA, ab124964, Abcam, Cambridge, UK), diluted 1:500 in PBS, was applied at 4°C overnight. Then, the fluorescent secondary antibody (diluted 1:300 in PBS) was added for 1 h at room temperature in darkness. DAPI (1:2000, Beyotime) was added to the cells and incubated for 10 min in darkness for nuclear staining. Fluorescence images were captured using a fluorescent microscope and quantified by ImageJ software. Each experiment was performed in triplicate.
5-ethynyl-2′-deoxyuridine (EdU) incorporation assay
A total of 2000 LX-2 cells were seeded in each well of 24-well plates. The EdU kit (Epizyme Biotech) was utilized to fix and stain the proliferating LX-2 cells according to the manufacturer's instructions. Hoechst was used to stain the cell nuclei in darkness. Samples were observed under a fluorescence microscope and then quantified using ImageJ software. Each experiment was performed in triplicate.
Transwell assay
To induce cell migration, 200 μL serum free medium with 2 × 104 LX-2 cells was seeded onto the upper chamber and 600 μL medium containing 10% FBS was added to the lower chamber. Cells were cultured at 37°C in a 5% CO2 atmosphere for 48 h and then the lower surface of the upper chamber was fixed with methanol for 10 min and stained with 0.5% crystal violet for 20 min. A light microscope was employed to observe the migrated cells. Each experiment was performed in triplicate.
Protein interaction analysis
The amino acid sequences of MEOX1 (Uniprot # P50221), PAI-1/SERPINE1 (Uniprot # P05121), and Smad2 (Uniprot # Q15796) were retrieved from Uniprot (https://www.uniprot.org/) and subsequently submitted to Alphafold3 (https://alphafoldserver.com/). To visualize the predicted protein interaction structures with high predictive local distance difference test and predictive template modeling scores, PyMOL (https://pymol.org/2/) was employed.
Statistical analysis
The analysis was carried out by R software (version 4.2.3), GraphPad Prism (version: 7.00) and SPSS software (version: 20.0). All data were presented as mean ± SEM. Statistical significance between groups was determined by one-way analysis of variance or Student t tests. Results were considered statistically significant with values of P < 0.05.
Results
Identification of DETFs in the MASH patients
To identify the DETFs related to MASH-related liver fibrosis in the Gene Expression Omnibus (GEO) database, we designed a flow chart (Figure S1(a)). Firstly, as shown in Figure S1(b), we detected the DEGs between the samples of hepatic steatosis patients and MASH patients within each of the GSE135251 and GSE167523 (false discovery rate < 0.05 and |log2 fold change| ≥ 1). We further screened transcription factors based on the consistently DEGs across both datasets to obtain key regulatory genes. Finally, 30 DETFs were identified and the Figure S1(c) suggested that the DETFs were significantly correlated with the progression of liver fibrosis in MASH patients.
WGCNA and identification of module associated with the stage of MASH-related liver fibrosis
To identify the genes that are synergistically expressed and associated with the stage of MASH-related liver fibrosis, we performed a WGCNA to determine the key module genes in MASH-related liver fibrosis. Firstly, we eliminated the visible outliers through sample clustering (Figure 1(a)). As shown in Figure 1(b) and (c), we selected soft-thresholding power β = 14 (scale-free R2 = 0.85) as the appropriate soft threshold by constructing a scale-free topology model and analyzing the mean connectivity of the adjacency. To identify the key modules associated with the stage of MASH-related fibrosis, we performed a cluster dendrogram and a dynamic tree-cutting (Figure 1(d)). Subsequently, a total of 20 co-expression modules were identified in WGCNA as shown in Figure 1(e), among which the dark red module exhibited the highest correlation with MASH-related liver fibrosis stage fibrosis-3 (F3) and fibrosis-4 (F4) compared to the other modules (P = 6.2E-4 and 2.2E-10, respectively; Figure 1(f)). Therefore, the dark red module, which contains 844 genes, was regarded as the MASH-related liver fibrosis stage F3 module (Figure 1(g)). Meanwhile, a total of 1033 genes contained in the dark red module were filtered according to the standard pipeline, which was associated with the MASH-related liver fibrosis stage F4 (Figure 1(h)).
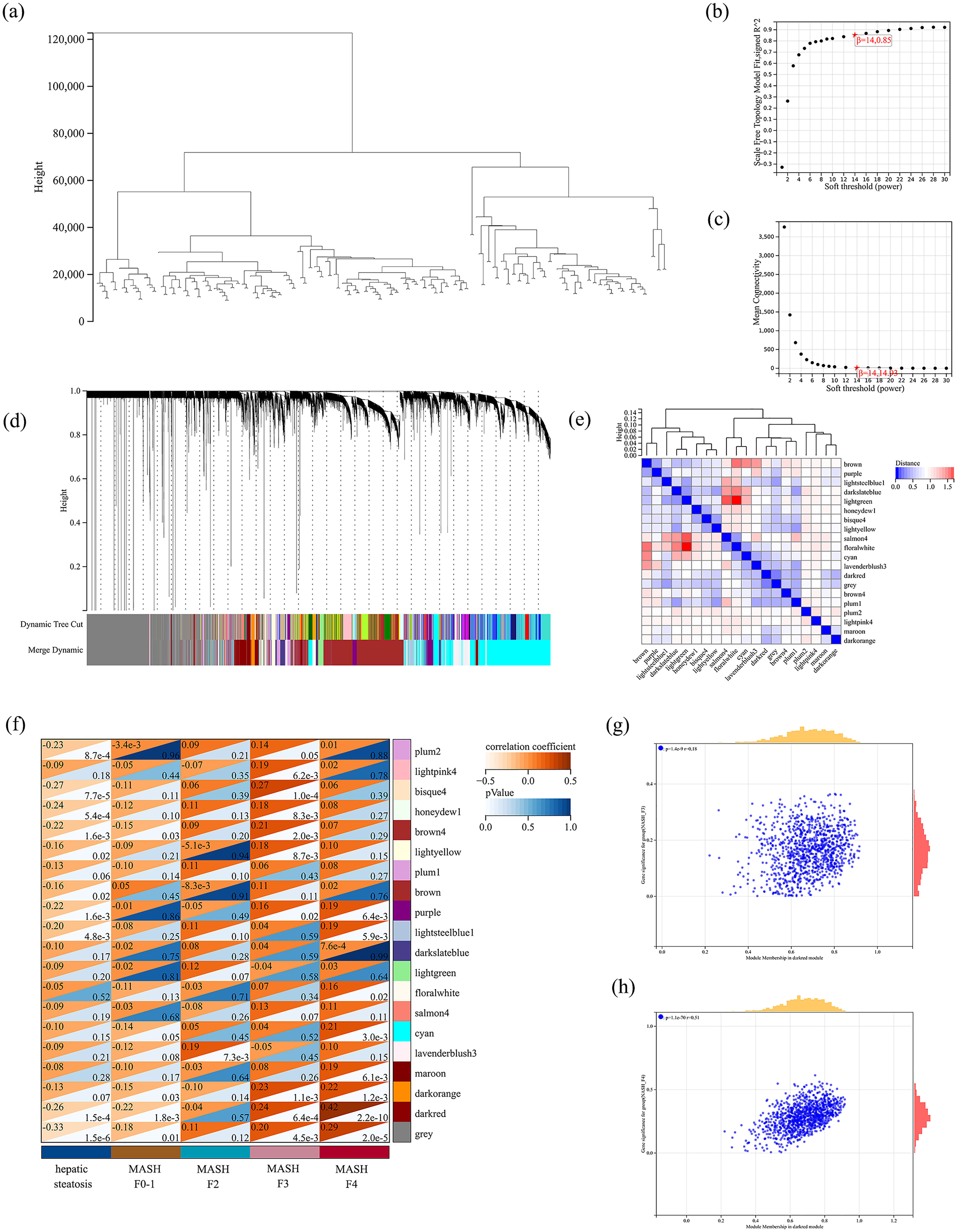
Construction of WGCNA for MASH-related liver fibrosis and identification of the hub module. (a) Clustering dendrogram of samples based on their Euclidean distance. (b) The scale-free index versus the soft-thresholding power. (c) The mean connectivity versus the soft-thresholding power. (d) The Cluster dendrogram of co-expression network modules from WGCNA, with dissimilarity based on topological overlap, together with assigned module colors. (e) Dendrogram of all DEGs clustered based on different similarity measures. (f) Heatmap depicting the correlation between module feature genes (MES) and the stage of MASH-related liver fibrosis. (g) Scatterplot correlation between the dark red module and MASH-related fibrosis stage F3. (h) Scatterplot correlation between the dark red module and MASH-related fibrosis stage F4.
Identification of DETFs that regulate the dark red module
Within the dark red module, DEGs related to MASH-related liver fibrosis stage F3 and F4 were both identified. Among these genes, a total of six DETFs (MEOX1, SOX4, LEF1, SOX9, MYC, and CBX2) were recognized as hub genes that were intricately linked to MASH-related liver fibrosis (Figure 2(a)). Subsequently, the analysis of the GEO database GSE135251 indicated that the expressions of the six DETFs in the liver tissue of MASH patients were significantly higher than that of hepatic steatosis patients, and were positively correlated with the stage of liver fibrosis (Figure 2(b)).
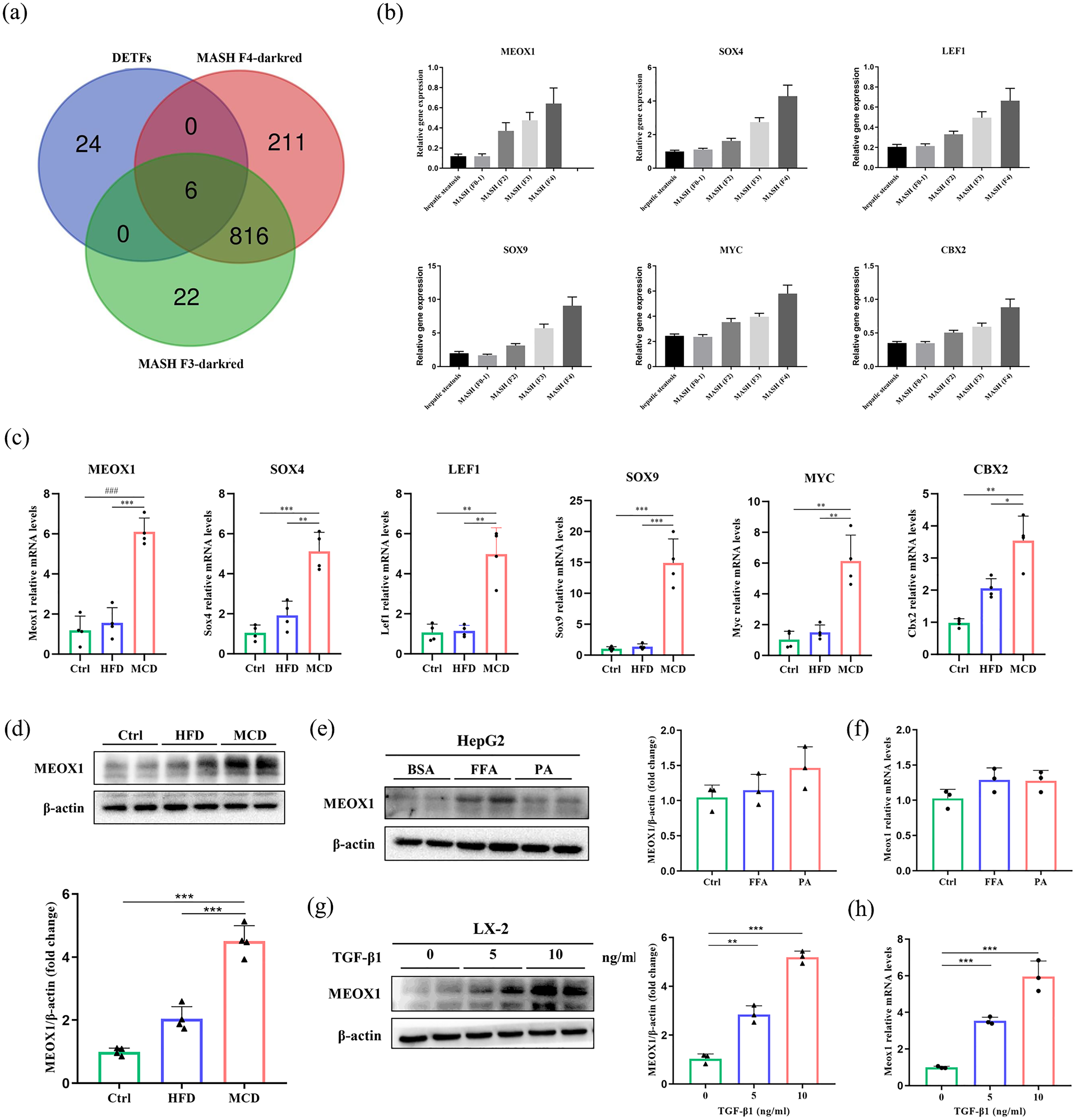
Identification and validation of DETFs that regulate the dark red module. (a) Venn diagram displaying DETFs linked to MASH-related liver fibrosis stage F3 and F4. (b) Gene expression levels of MEOX1, SOX4, LEF1, SOX9, MYC, and CBX2 across varying stages of MASH-related liver fibrosis in GSE135251. (c) Representative mRNA expressions of MEOX1, SOX4, LEF1, SOX9, MYC, and CBX2 measured in chow-fed mice, HFD-fed mice, and MCD-fed mice (n = 4/group). (d) Representative protein levels of MEOX1 measured in chow-fed mice, HFD-fed mice, and MCD-fed mice (n = 4/group). (e) Representative protein levels and (f) mRNA levels of MEOX1 measured in FFA and PA treated HepG2 cell (n = 3/group). (g) Representative protein levels and (h) mRNA levels of MEOX1 measured in a TGF-β1-treated LX-2 cell (n = 3/group). *P < 0.05; **P < 0.01; ***P < 0.001; ### P < 0.0001.
Validation of DETFs that regulate the dark red module
Next, to verify the expression of DETFs in vivo, HFD and MCD diets were employed to establish the models of hepatic steatosis and MASH-related liver fibrosis, respectively. As shown in Figure S2, compared to the control group, histological analysis using H&E staining and Masson staining revealed marked enhancement of lipid deposition in the HFD group and liver fibrosis in the MCD group. Additionally, serum ALT, AST, and TG levels were significantly higher in HFD and MCD mice, indicating improved liver damage.
In comparison with chow-fed mice, the mRNA levels of the six DETFs were not significantly altered in the HFD group, but were markedly increased in model of MASH-related fibrosis (Figure 2(c)). MEOX1 was selected for further validation for the following reasons: (a) The role of MEOX1 in MASH-related fibrosis was largely undefined; (b) MEOX1 has attracted increasing attention in recent studies as a novel biomarker for myocardial fibrosis and heart failure22,23; and (c) As a transcription factor, the clinical value and molecular mechanism of MEOX1 deserve further exploration due to the scarcity of relevant studies.
In line with the qRT-PCR result, we detected no significant elevation in MEOX1 protein levels in the HFD-induced mice compared with the control group through Western blot assay, while we observed the changes of MEOX1 in the livers of MCD-induced MASH-related fibrosis mice (Figure 2(d)). To determine the potential role of MEOX1 in the pathogenesis of MASH-related liver fibrosis, we established hepatocyte models of MASH by treating HepG2 with FFA and PA for 24 h, and TGF-β1 was used to stimulate the LX-2 cells for 24 h to construct an activated HSCs model of MASH-related fibrosis. The results of Western blot and qRT-PCR indicated that the expression level of MEOX1 was not significantly altered by FFA and PA stimulation (Figure 2(e) and (f)). Conversely, Figure 2(g) and (h) revealed that the expression of MEOX1 was substantially increased in activated HSCs compared to quiescent HSCs. The above results suggest that MEOX1 might play a promoting role in fibrosis progression and HSCs activation in MASH patients.
MEOX1 regulates HSCs activation, proliferation, and migration
To further determine the functional role of MEOX1 in MASH-related liver fibrosis in vitro, we constructed MEOX1 knockdown models using siRNAs (siMEOX1). We observed that the expression of MEOX1 protein and mRNA levels in the siMEOX1 + PA + HepG2 group and the siMEOX1 + TGF-β1 + LX-2 group were significantly decreased compared to the controls. Moreover, we discovered that compared to the siCtrl + PA + HepG2 group, the expression level of fatty acid uptake and synthesis- related gene fatty acid synthase (FASN) was not significantly reduced (Figure 3(a)). Meanwhile, MEOX1 silencing did not notably reduce PA-induced lipid deposition compared with the control group (Figure 3(b)). In contrast, as shown in Figure 3(c) and (d), the myofibroblast marker gene α-smooth muscle actin (α-SMA) was down-regulated in the siMEOX1 + TGF-β1 + LX-2 group compared to the siCtrl + TGF-β1 + LX-2 group. Notably, the results of the EdU assay (Figure 3(e)) and Transwell assay (Figure 3(f)) indicated that MEOX1 knockdown significantly reduced the proliferation and migration of activated HSCs. Taken together, these findings establish MEOX1 as a crucial regulator of HSCs activation, proliferation, and migration rather than hepatocyte lipid deposition.
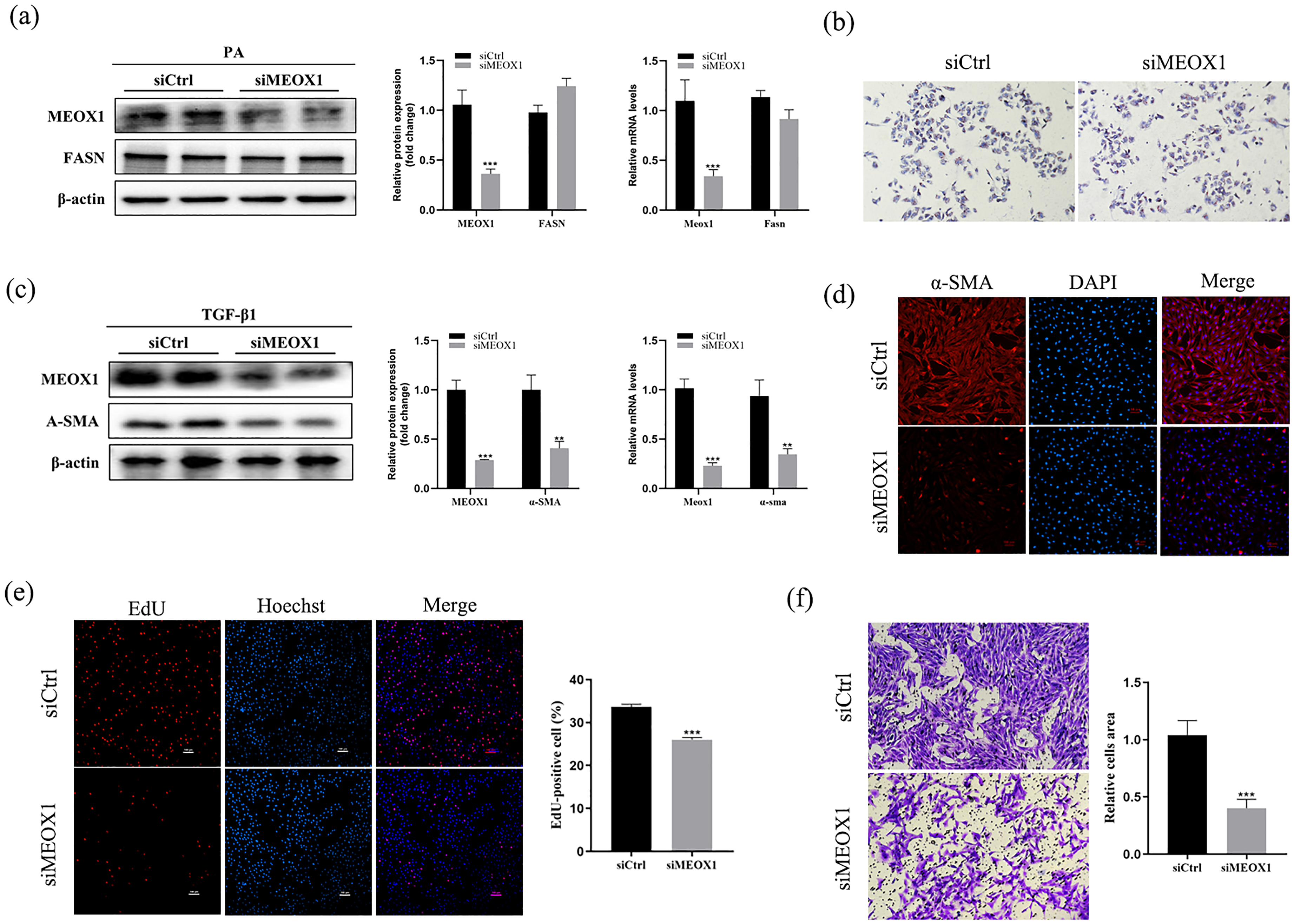
MEOX1 regulates HSCs activation, proliferation, and migration. (a) Western blot and qRT-PCR analysis of MEOX1 and FASN expression in HepG2 cells transfected with siCtrl/ siMEOX1 and stimulated with PA. (b) Oil O red staining of HepG2 cells transfected with siCtrl/ siMEOX1 and stimulated with PA. (c) Western blot and qRT-PCR analysis of MEOX1 and α-SMA expression in LX-2 cells transfected with siCtrl/ siMEOX1 and stimulated with TGF-β1. (d) Immunocytofluorescence staining of α-SMA in LX-2 cells transfected with siCtrl/ siMEOX1 and stimulated with TGF-β1. (e) EdU assay in LX-2 cells transfected with siCtrl/ siMEOX1 and stimulated with TGF-β1. (f) Transwell assay in LX-2 cells transfected with siCtrl/ siMEOX1 and stimulated with TGF-β1. All data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. All images are representative of three independent experiments.
SERPINE1 is a downstream target transactivated by MEOX1 during HSCs activation
To further determine the downstream mechanism of MEOX1 in HSCs activation, we performed RNA-seq of LX-2 cells with MEOX1 knockdown and control groups stimulated with TGF-β1 (Figure 4(a)). Rely on the sequencing data, a total of 365 DEGs were identified (adjusted P value < 0.05 and fold change > 1.5, Figure 4(b)), among which 152 DEGs were up-regulated and 213 were down-regulated (Figure 4(c)). Further GO analysis revealed major enrichment in ECM, wound healing, and cell proliferation, which were closely related to the progression of liver fibrosis (Figure 4(d)). The results of Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis indicated that hypoxia inducible factor-1 (HIF-1) signaling pathway, an important signaling pathway involved in the pathogenesis of MASH and liver fibrosis, 24 was among the top 10 enriched pathway regulated by MEOX1 during MASH-related liver fibrosis (Figure 4(e)). Consistently, Gene Set Enrichment Analysis (GSEA) showed that HIF-1 signaling pathway was involved in the regulation of MEOX1 on liver fibrosis (|NES| = |-1.54|, P = 0.007, FDR =0.385, Figure 4(f)). Then, we further explored the potential downstream target of MEOX1 by analyzing DEGs enriched in the HIF-1 pathway (Figure 4(g)). A total of 10 down-regulated DEGs (SERPINE1, HMOX1, NOS2, SLC2A1, ENO2, EGLN1, CDKN1B, PDK1, PGK1, and VEGFA) were identified as the potential targets of MEOX1 (Figure 4(h)). Furthermore, we constructed a STRING network to identify the ten hub genes that were central to signaling pathway (Figure 4(i)), and found that SERPINE1, as the most significant DEGs related to HIF-1 pathway regulated by MEOX1, could interact with multiple proteins (Figure 4(j)). Collectively, these results suggested that SERPINE1 was a downstream target transactivated by MEOX1 during HSCs activation and MASH-related liver fibrosis.
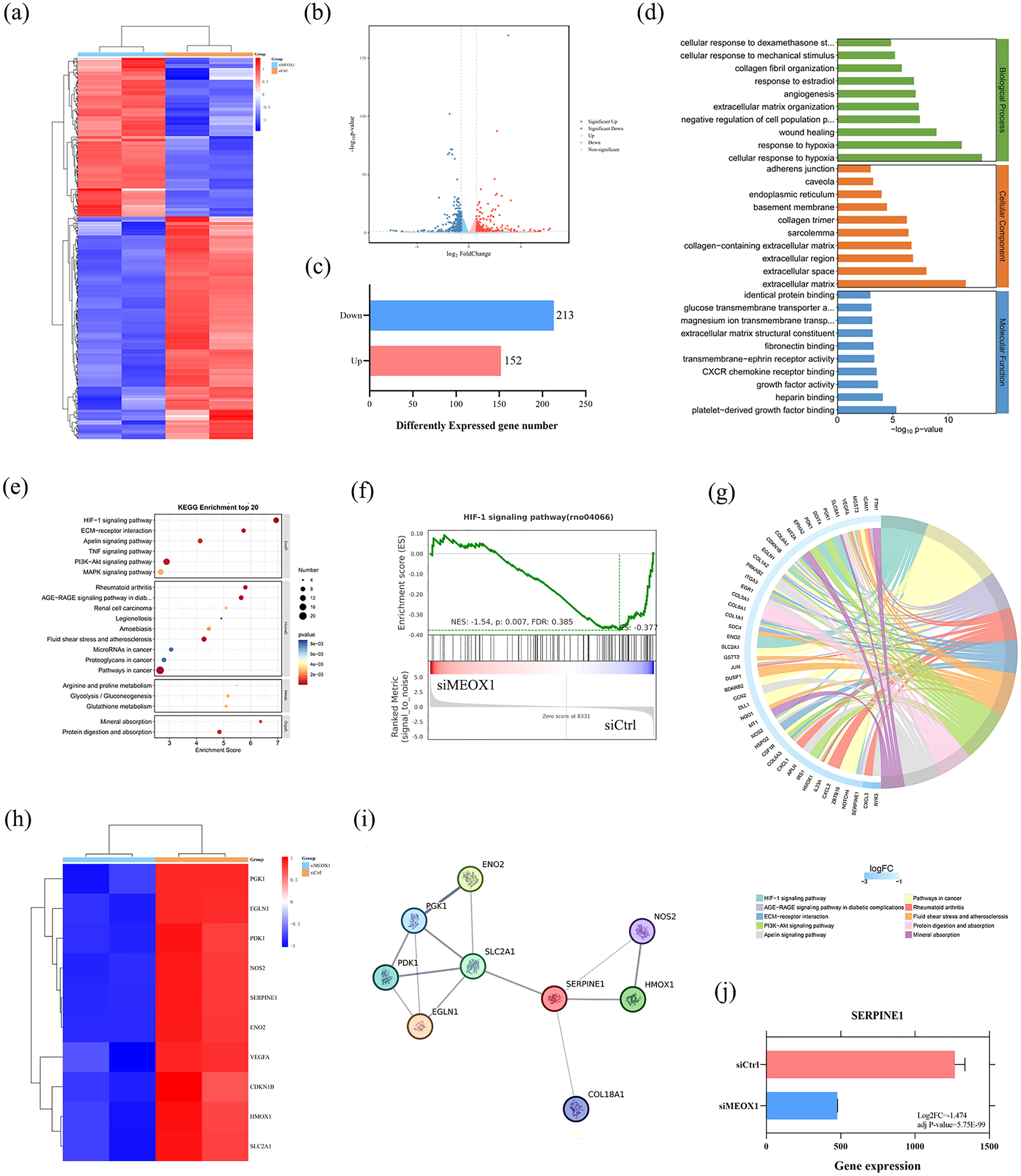
Exploration of the potential downstream target of MEOX1 in HSCs. (a) Heatmap of DEGs between MEOX1 knockdown LX-2 cells and control groups stimulated with TGF-β1 identified from RNA-seq. (b) Volcano plot of RNA-seq data. (c) The number of up-regulated and down-regulated DEGs. (d) The GO enrichment analysis of DEGs. (e) The KEGG enrichment analysis of DEGs. (f) GSEA plot of the regulation of HIF-1 signaling pathway based on the RNA-seq data. (g) The KEGG enrichment analysis of the hub genes related to the top 10 pathways. (h) Heatmap of hub down-regulated DEGs enriched in HIF-1 signaling pathway. (i) STRING network of the hub down-regulated DEGs. (j) Gene expression of SERPINE1 in the RNA-seq data.
Smad2/MEOX1/SERPINE1 pathway is responsible for regulation of MASH-related fibrosis
By predicting the binding site of MEOX1 and SERPINE1 using the JASPAR database (jaspar.genereg.net/), we found that MEOX1 might bind to the promoter of SERPINE1 gene (Figure 5(a)). The structure of the protein complex formed by the MEOX1 and SERPINE1 were predicted with the latest protein complex structure prediction software Alphafold-Multimer released by the DeepMind team (Figure 5(c) and (d)). 25 Further visual analysis disclosed the existence of protein interaction sites between MEOX1 and SERPINE1 (Figure 5(e)). Through analyzing the binding interface structures and the binding site predicted by the JASPAR database, our data indicated that SERPINE1 was a downstream target transactivated by MEOX1 during the progression of MASH-related liver fibrosis. Meanwhile, as the most critical pathway in liver fibrosis, consistent with the above predictions, we identified that the TGF-β1/Smad2 signaling cascade acts as an upstream regulator of MEOX1, modulating its expression (Figure 5(b) and (d)). The results showed that Smad2/MEOX1/SERPINE1 pathway was accountable for the regulation of MASH-related fibrosis, and further in-depth exploration was warranted.
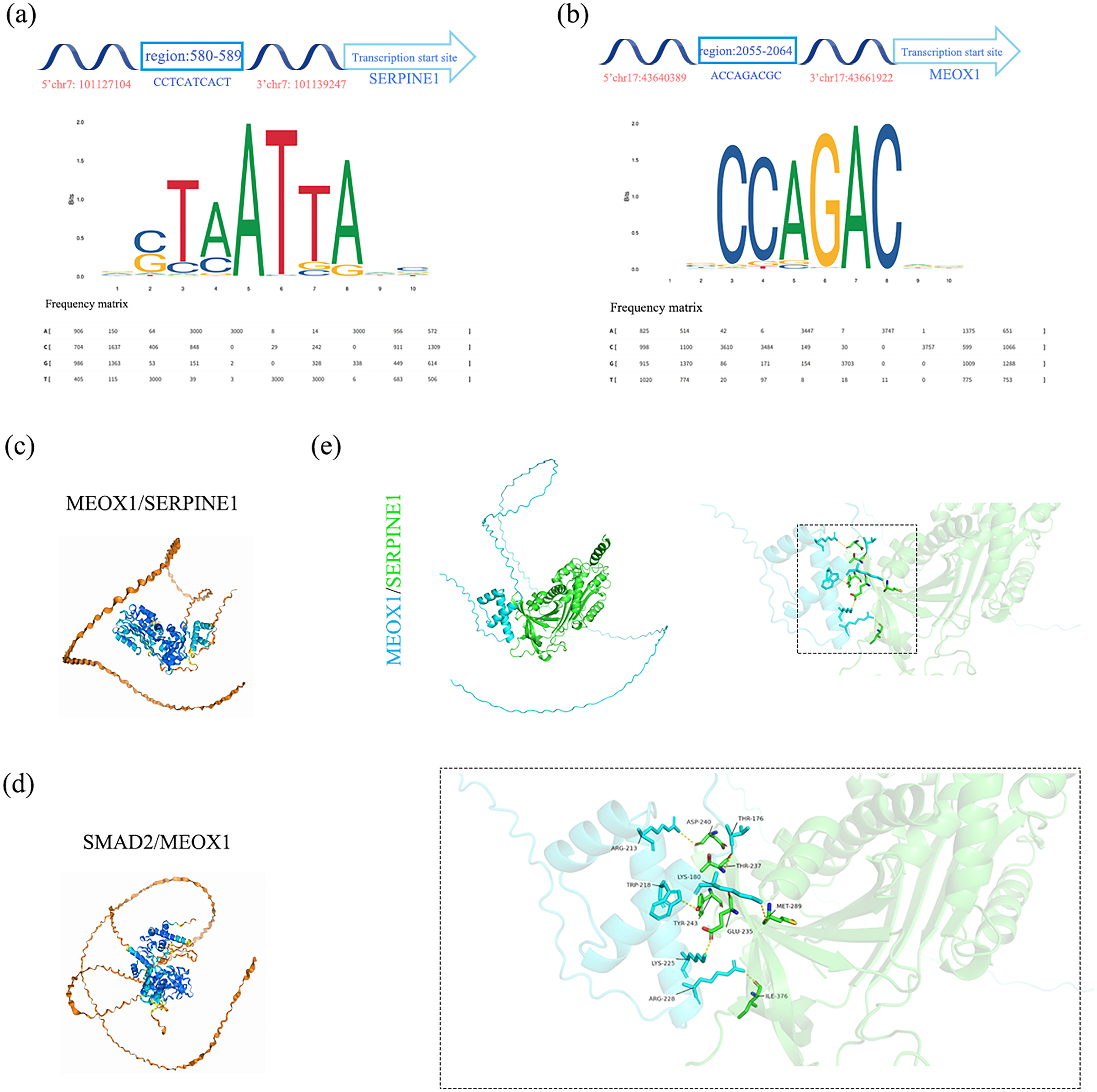
Smad2/MEOX1/SERPINE1 pathway was responsible for regulation of MASH-related fibrosis. (a) Top one binding region of MEOX1 in the promoter sequence of SERPINE1. (b) Top one binding region of Smad2 in the promoter sequence of MEOX1. (c) Protein interaction structure of MEOX1/SERPINE1 predicted by Alphafold 3. (d) Protein interaction structure of Smad2/MEOX1 predicted by Alphafold 3. (e) Protein interaction structure of MEOX1/SERPINE1 visualized by PyMOL.
Discussion
As the major determinant of disease prognosis, liver fibrosis plays a crucial role in the development of MASLD from simple steatosis to steatohepatitis and serves as an independent risk factor for poor prognosis (including cirrhosis, liver cancer, and cardiovascular events).5–7 Hence, anti-fibrotic therapy constitutes an important strategy in the development of drugs for MASH. 26 The excessive activation of HSCs is the core link in the progression from liver steatosis to fibrosis in patients with MASLD. 10 Persistent fibrotic stimulation results in excessive activation of HSCs, which in turn causes an imbalance in the deposition and degradation of ECM, thereby accelerating the progression of fibrosis. 27
The rapid advancement of bioinformatics methods has facilitated an increasing number of researchers to identify the latest molecular biomarkers for disease diagnosis and treatment by means of data mining and analysis of sequencing data or gene chips. 28 In our study, by analyzing DEGs and DETFs associated with MASH-related fibrosis from the GEO database, constructing a co-expression network and identifying the different modules via WGCNA, we discovered six DETFs (MEOX1, SOX4, LEF1, SOX9, MYC, and CBX2) associated with the stage of MASH-related liver fibrosis. Subsequently, through gene expression analysis in GSE135251 and qRT-PCR assay, we found that the expressions of the six hub DETFs were upregulated in MASH and were strongly correlated with the stage of MASH-related liver fibrosis.
First identified in the developing somitic mesoderm, transcription factor MEOX1 was mainly localized to the nucleolus, and played a crucial role in cell differentiation, migration, and proliferation. 29 Evidence suggests that aberrant expressions of MEOX1 are associated with a broad spectrum of pathologies, covering both non-neoplastic conditions, such as skeletal disorders, cardiovascular diseases, and Crohn's disease, as well as neoplastic diseases, including breast cancer, ovarian cancer, and intrahepatic cholangiocarcinoma.30,31 Notably, MEOX1 is strongly associated with fibroblast activation induced by cardiac remodeling during myocardial fibrosis.22,23 Meanwhile, silencing MEOX1 markedly inhibited the progression of pulmonary fibrosis in mice. 32 These findings imply that MEOX1 may also have a potential value in HSCs activation. However, whether MEOX1 participates in liver fibrosis and the underlying mechanisms remain unclear. To confirm the enhanced expression of MEOX1 in MASH-related liver fibrosis, we quantified the MEOX1 levels in HepG2 stimulated by FFA/PA and HSCs stimulated by TGF-β1. Here we discovered that MEOX1 was significantly upregulated in activated HSCs, but not in high-fat hepatocyte model. Furthermore, to explore the biological functions of MEOX1 in the progression of MASH-related fibrosis, we knocked down MEOX1 expression by transfection with siRNAs in HepG2 and LX-2 cell lines and found that down-regulated MEOX1 expression markedly suppressed the activation, proliferation, and migration of HSCs. In contrast, silencing MEOX1 did not exert an inhibitory effect on lipid deposition in hepatocyte. These data support the pivotal role of MEOX1 in HSCs activation and liver fibrosis progression.
To identify the downstream targets of MEOX1 transcriptional activation, we employed RNA-seq to analyze the expression profiles of HSCs exhibiting a low expression of MEOX1 relative to the control group. As a crucial signaling pathway involved in the progression of steatosis, inflammation, fibrosis, and vascular dysfunction in MASH, the HIF-1 signaling pathway was prominently highlighted by both KEGG enrichment analysis and GSEA. 33 Through conducting a STRING network of DEGs related to the HIF-I signaling pathway, we found that SERPINE1 is a vital target gene. As a cancer-promoting factor, SERPINE1 exhibits elevated expression levels in various tumors, contributing to tumor cell proliferation, migration, invasion, and angiogenesis. 34 In support, Kim et al. 12 have reported that SERPINE1 (PAI-1) may regulate the activation of HSCs through integrin αv-dependent activation of latent TGF-β. The deletion of SERPINE1 in HSCs or the inhibition of PAI-1 significantly attenuated the progression of MASH-related liver fibrosis. Our study utilized JASPER, AlphaFold, and PyMOL to predict that MEOX1 has the potential to regulate the SERPINE1 promoter region.
TGF-β1 serves as a principal pathogenic factor and “key regulator” in liver fibrosis, and its overexpression can induce the onset and progression of various fibrotic diseases. 35 Nevertheless, inhibiting TGF-β1 expression carries the risk of exacerbating autoimmune diseases. 36 Therefore, exploring new targets within the downstream pathways of TGF-β1 is of vital importance for the effective treatment of liver fibrosis. 37 Smad2, functioning as an intracellular mediator of the TGF-β1 signaling pathway, mediates the majority of the profibrotic activities of TGF-β1. 36 The study conducted by Alexanian et al. 22 demonstrated that Smad2 serves as a transcriptional mediator in TGF-β1 signal transduction pathways, and the knockdown of Smad2 results in diminished MEOX1 transcription levels, a finding corroborated by the results from the predictions of JASPER and AlphaFold.
To date, an extensive body of research has confirmed that cellular networks comprising hepatocytes, macrophages, and HSCs are the primary drivers of MASH-associated liver fibrosis. In subsequent studies, we aim to further investigate the role and mechanism of MEOX1 in cellular interactions, thereby providing a more robust theoretical and experimental foundation for the development of MEOX1-targeted therapeutics.
Conclusions
In conclusion, we identify MEOX1 as a potential molecular marker for the diagnosis and treatment of MASH-related liver fibrosis, which is correlated with the stage of fibrosis. This is the first report to demonstrate that the expression level of MEOX1 is correlated with the stage of MASH-related liver fibrosis through the upregulation of SERPINE1 expression and the mediation of the fibrosis-promoting effects of the TGF-β1/Smad2 pathway.
Supplemental Material
sj-docx-1-jbm-10.1177_03936155251335975 - Supplemental material for Identification of MEOX1 as a potential target in metabolic dysfunction-associated steatohepatitis-related liver fibrosis
Supplemental material, sj-docx-1-jbm-10.1177_03936155251335975 for Identification of MEOX1 as a potential target in metabolic dysfunction-associated steatohepatitis-related liver fibrosis by Xiaoxiao Jiao, Linying Lai, Yiting Qian, Bo Sun and Wenzhuo Yang in The International Journal of Biological Markers
Footnotes
Ethical consideration
Our study was approved by the Animal Ethics Committee of Tongji University (approval no. TJHBLAC-2019-024).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant numbers 81873567, 82370579).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.