
Abstract
Fibrosis accumulation is a dynamic process resulting from a wound-healing response to acute or chronic liver injury of all causes. The cascade starts with hepatocyte necrosis and apoptosis, which instigate inflammatory signaling by chemokines and cytokines, recruitment of immune cell populations, and activation of fibrogenic cells, culminating in the deposition of extracellular matrix. These key elements, along with pathways of transcriptional and epigenetic regulation, represent fertile therapeutic targets. New therapies include drugs specifically designed as antifibrotics, as well as drugs already available with well-established safety profiles, whose mechanism of action may also be antifibrotic. At the same time, the development of noninvasive fibrogenic markers, and techniques (e.g. fibroscan), as well as combined scoring systems incorporating serum and clinical features will allow improved assessment of therapy response. In aggregate, the advances in the elucidation of the biology of fibrosis, combined with improved technologies for assessment will provide a comprehensive framework for design of antifibrotics and their analysis in well-designed clinical trials. These efforts may ultimately yield success in halting the progression of, or reversing, liver fibrosis.
Introduction
Liver fibrosis and its end-stage sequela of cirrhosis resulting from chronic liver injury are major causes of morbidity and mortality worldwide. Among the etiologies of hepatic fibrosis, viral infection is most common (e.g. hepatitis B and C), and currently affects 1–2% (5.3 million) of the US population [Mitchell et al. 2010], with cirrhosis projected to reach 45% of those infected with hepatitis C virus (HCV) patients in 2030 [Davis et al. 2010]. In addition, the consequences of precipitously rising obesity rates worldwide have accelerated the risk of liver injury due to nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH); at least 20% of patients with NASH progress to cirrhosis [Edmison and McCullough, 2007]. Other etiologies of chronic liver injury include alcohol-induced disease, drug-induced toxicity, other liver infections (e.g. schistosomiasis), immune-mediated liver diseases (e.g. autoimmune hepatitis), metabolic disorders (e.g. lipid, glycogen, or metal storage disorders) and cholestasis (e.g. secondary biliary cirrhosis). Complications of cirrhosis are well characterized, and include ascites, portal hypertension, encephalopathy, liver failure, and accelerated risk of hepatocellular carcinoma (HCC). In particular, HCC has the fastest rising cancer incidence of any neoplasm in the USA and Europe [Jemal et al. 2009].
At present, the only curative treatment for end-stage cirrhosis is transplantation [Said and Lucey, 2008]. Even in the developed world, however, the number of donor organs available and the condition of the potential recipients limit the applicability of this technique, and thus new medical therapies to halt or reverse fibrosis are urgently needed.
The discovery of the main fibrogenic cell of injured liver, the activated hepatic stellate cell (HSC)/myofibroblast, and the compelling evidence that fibrosis is a dynamic and even reversible process, have spurred efforts to uncover the molecular mechanisms of fibrosis in order to expedite new therapeutic strategies. Several scientific advances have presaged this progress. First, the development of cell isolation and culture techniques for HSCs from rodents and humans [Friedman et al. 1992] was a major advance, in particular the recognition that growth of primary HSCs on plastic mimicked their response to liver injury in vivo. Second, the development of animal models of liver injury due to hepatic toxins (e.g. CCl4, thioacetamide, dimethylnitrosamine), bile duct ligation, and/or immune injury (e.g. concanavalin A) have helped unearth key fibrogenic mediators. Third, genetic models of liver disease generated by gene targeting in mice have revealed key determinants of experimental hepatic fibrosis. Fourth, the development of powerful techniques to study transcriptomic expression profiles has shed new light on genes that are up- and down-regulated in both activated HSCs and the fibrotic liver [Bataller et al. 2003a]. Finally, contributions of microRNAs and epigenetic signaling to fibrogenesis have been uncovered recently, and their impact on fibrosis and gene regulation are increasingly appreciated. For example, a recent gene array analysis identified downregulation of the miR-29-family in mouse models of fibrosis, which parallels findings in patients with more advanced hepatic fibrosis. Downregulation of miR-29 in HSCs is mediated by transforming growth factor (TGF)-β, lipopolysaccharide (LPS), and nuclear factor (NF)-κB. In addition, overexpressing miR-29b in HSCs lead to a decrease in collagen production [Roderburg et al. 2011]. In aggregate, as these pathways of fibrogenesis are further clarified, the opportunity to develop new diagnostic tools and therapeutic strategies will accelerate.
Cellular and molecular pathogenesis of liver fibrosis
Figure 1 provides a schematic illustration of fibrosis progression.
Schematic illustration of fibrosis progression. Hepatocyte damage and injury generate necrosis and apoptosis, which provoke cellular changes that stimulate the recruitment of inflammatory cells and activate fibrogenic cells. Different secreted signals from resident and recruited fibrogenic cells promote extracellular matrix synthesis. Repetitive bouts of injury trigger further progression to cirrhosis, whereas if injury subsides, then fibrosis regression may follow. Illustration courtesy of Alessandro Baliani, Copyright © 2011.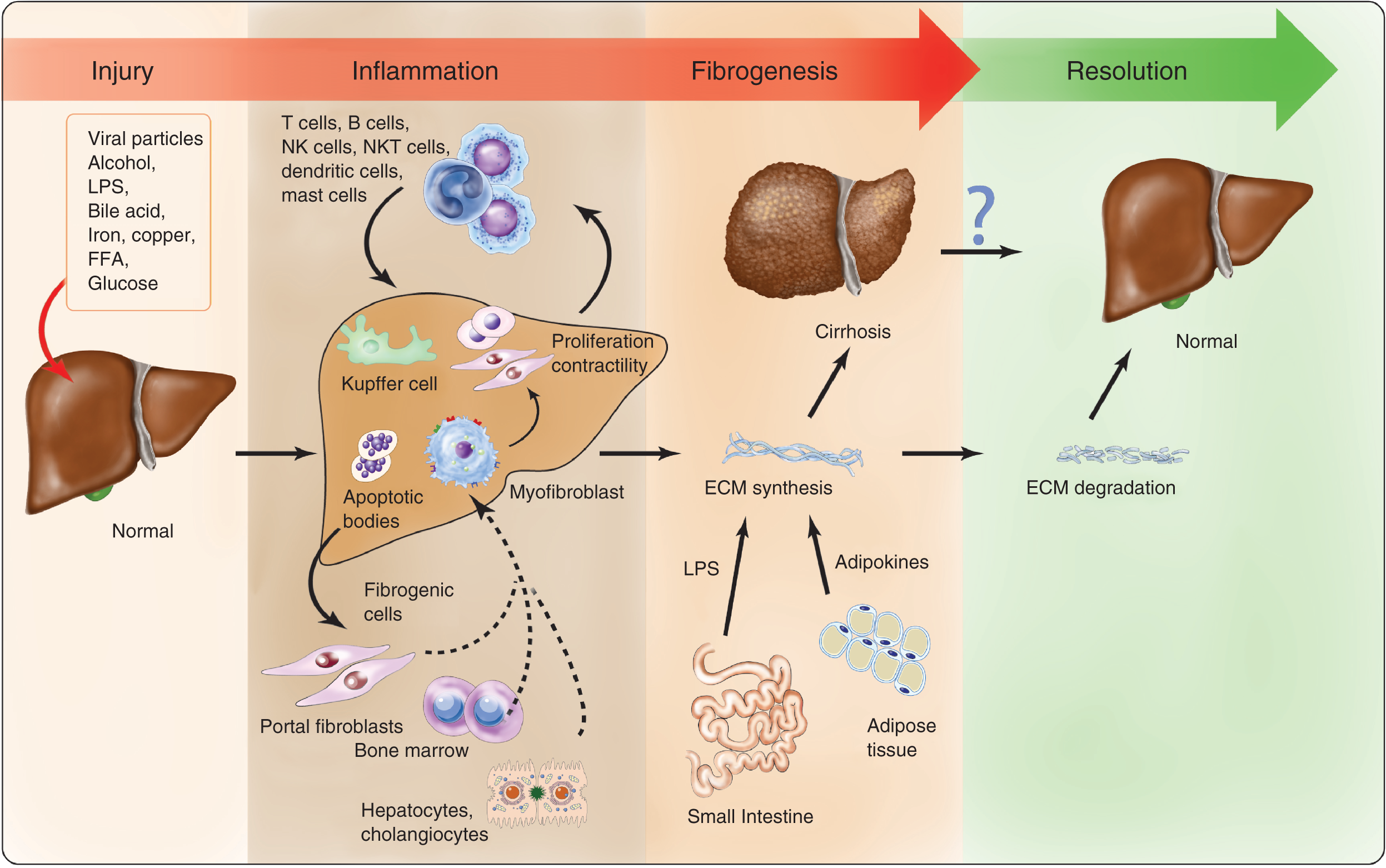
Initiation of injury signaling
Liver injury of all causes typically induces hepatocyte necrosis and apoptosis. Necrosis engages classic inflammatory and fibrogenic signals, and more recent findings suggest that apoptosis is also fibrogenic by releasing hepatocyte apoptotic bodies that are cleared through phagocytosis by Kupffer cells and HSCs (Figure 2). Engulfment of apoptotic bodies by HSCs triggers a profibrogenic response that stimulates TGF-β1 expression which induces collagen I, a major constituent of the cirrhotic scar [Canbay et al. 2002]. Moreover, phagocytosis of apoptotic bodies by quiescent HSCs facilitates their phenotypic transformation to myofibroblasts [Canbay et al. 2002]. Engulfment of apoptotic bodies by Kupffer cells enhances the expression of pro-fibrogenic genes (e.g. TGF-β1) and of death ligands (e.g. FasL), which initiate independent intracellular signaling cascades that further amplify liver injury [Canbay et al. 2004]. Damaged hepatocytes and phagocytic Kupffer cells both release reactive oxygen species (ROSs), as well as chemokines and fibrogenic and inflammatory mediators that recruit inflammatory cells and provoke HSC activation.
The inflammatory cascade. The damaged hepatocyte releases apoptotic bodies which are engulfed by resident Kupffer cells and hepatic stellate cells (HSCs), which then promote HSC activation. Paracrine stimulation by reactive oxygen species, chemokines and cytokines also promote HSC activation. Chemokines either from the injured cells or activated HSCs also stimulate the recruitment of inflammatory cells that augment this cascade. Illustration courtesy of Alessandro Baliani, Copyright © 2011.
Cellular sources of fibrinogenesis: role of the hepatic stellate cell
Scar formation is a common host response to repeated tissue injury. The prototypical fibrogenic cell in tissue repair is the contractile, proliferating and fibrogenic myofibroblast that expresses α-smooth muscle actin (α-SMA). The major source of myofibroblasts in liver injury is the HSC, a mesenchymal vitamin A-rich cell located in the subendothelial space of Disse, between hepatocytes and sinusoidal endothelial cells [Friedman, 2008a]. Recognition of HSCs as vital collagen-producing cells following their ‘activation’ into myofibroblasts during liver injury was a key advance in understanding the fibrotic response in liver [Friedman, 2004a; Friedman et al. 1985].
Activation of HSCs can be divided into two phases: initiation and perpetuation [Friedman, 2008b]. Initiation, also called a ‘pre-inflammatory’ stage refers to early changes in gene expression that result primarily from paracrine stimuli derived from damaged resident liver cells (sinusoidal endothelial cells, Kupffer cells, hepatocytes) and platelets. Kupffer cell engagement drives release of cytokines (especially TGF-β) and ROS signaling (see above) [Bilzer et al. 2006]. Endothelial cells participate in the conversion of latent TGF-β into the active form and produce fibronectin, which also provokes early HSC activation. In addition, platelets produce platelet-derived growth factor (PDGF), TGF-β and endothelial growth factor (EGF) which are potent activators of HSCs [Bachem et al. 1989].
Persistence of these stimuli accompanying sustained injury leads to a perpetuation phase regulated by autocrine and paracrine stimuli. Perpetuation involves at least seven distinct changes in HSC behavior, including proliferation, chemotaxis, fibrogenesis, contractility, altered matrix degradation, retinoid loss, and inflammatory signaling [Ghiassi-Nejad and Friedman, 2008]. As insights regarding the contributions of cytokine and chemokine signaling become clearer, new potential therapeutic targets emerge.
Alternative cellular sources of myofibroblasts
While HSCs remain the primary source of myofibroblasts, it is now clear that other mesenchymal cells also contribute to the myofibroblast population, including portal fibroblasts, bone marrow (BM)-derived cells, and possibly epithelial–mesenchymal transition (EMT) (Figure 1). The recruitment of these different fibroblasts populations is potentially disease-specific [Guyot et al. 2006].
Portal fibroblasts, because of their location within the portal areas, are recruited and activated into myofibroblasts primarily in biliary or cholestatic liver diseases [Kinnman and Housset, 2002]. Some of their signaling responses differ from HSCs, which could enable development of disease-specific antifibrotic therapies targeting this mesenchymal subpopulation [Dranoff and Wells, 2010].
A number of studies have implicated bone marrow cells as precursors of both myofibroblasts [Forbes et al. 2004] and circulating fibrocytes [Kisseleva et al. 2006]. Based on the same principle, the ability of BM-derived progenitor cells to migrate to the liver where they differentiate into fibrogenic or fibrolytic cells has provoked attempts to use autologous BM transplantation as a potential treatment to promote fibrosis reversion [Terai et al. 2006].
Epithelial cells can contribute to the replacement of dead or damaged hepatic cells through EMT. After liver injury, the most important trigger to EMT is the release of chemokines, matrix metalloproteinases (MMPs), and growth factors such as PDGF and TGF-β, via both the Smad2/3 and mitogen-activated protein kinase (MAPK)-dependent pathways. Recently, hedgehog signaling has been implicated in this process as well. Moreover, the transition could be bidirectional, mesenchymal–epithelium transition (MET) [Choi and Diehl, 2009].
Based on more recent studies, however, the contribution of EMT to fibrogenic cells in liver injury is probably minor. The use of transgenic mice in which marker genes are expressed in a manner that reflects the original tissue of origin has challenged the evidence that hepatocytes undergo EMT and deposit collagen in the injured liver [Taura et al. 2010]. Specifically, this system ‘marks’ cells with a reporter protein if they ever expressed an hepatocyte gene, even following their transition into a mesenchymal cell. Yet, such marked cells are rarely if ever seen in injury models. Evidence that cholangiocytes convert into mesenchymal cells using such an approach has been equally inconclusive [Scholten et al. 2010; Wells, 2010].
The immune system
The immune system plays an important role in fibrosis, since persistent inflammation almost always precedes and accompanies fibrosis. Both the innate and the adaptive immune systems contribute to the process. Damaged hepatocytes release apoptotic bodies, ROS cytokines (tumor necrosis factor [TNF]α, vascular endothelial growth factor [VEGF], insulin-like growth factor [IGF]-1), and chemokines (CXC) that activate endothelial cells, HSCs, and Kupffer cells by paracrine stimulation. These signals may lead to recruitment and activation of the inflammatory system (Figure 2).
Kupffer cells (liver macrophages) can activate HSCs and recruit inflammatory cells through secretion of ROS, profibrogenic cytokines, and chemokines, as well as prostanoids, nitric oxide, PDGF, TGF-β1, and monocyte chemotactic protein (MCP)-1. Blockade of macrophage infiltration into the site of injury prevents the transdifferentiation of HSCs to myofibroblasts and suppresses fibrosis [Imamura et al. 2005].
Similarly, activated HSCs increase the secretion of inflammatory chemokines (MCP-1, MCP-2, interleukin [IL1]-B) [Matsuoka and Tsukamoto, 1990], express adhesion molecules (e.g. intercellular adhesion molecule [ICAM]-I) [Hellerbrand et al. 1996], and modulate the immune system by functioning as antigen-presenting cells (APCs) to T cells and natural killer T (NKT) cells [Winau et al. 2007]. A positive feedback loop exists between inflammatory cells and HSCs. This includes T lymphocytes and especially, Th2 cells, which can also induce direct fibrogenic responses [Safadi et al. 2004].
Binding of bacterial LPS to the TLR4 receptor, which is expressed by HSCs and macrophages [Guo et al. 2009], stimulates production of ROS via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [Wheeler et al. 2001]. TLR4 also enhances TGF-β signaling via downregulation of a TGF-β pseudoreceptor, BAMBI [Seki et al. 2007]. The development of a TLR4 neutralizing agent is a therapeutic possibility based on studies using a TLR4/MD2 fusion protein, which inhibits LPS-induced pro-inflammatory signaling in HSCs [Schnabl et al. 2008]. In addition, apoptotic bodies from injured hepatocytes can repress HSC migration and increase collagen production [Watanabe et al. 2007]. TLR9-deficient mice display decreased hepatic fibrosis [Mencin et al. 2009]. Furthermore, signaling by TLR4 in myofibroblasts leads to activation of NF-κB, a pro-inflammatory transcription factor [Elsharkawy and Mann, 2007] that promotes cell survival (see below).
New evidence suggests that natural killer (NK) cells, a vital component of the innate immune system, promote resolution of the wound-healing process by clearing early activated, and senescent HSCs (see the Resolution of fibrosis section below) through production of interferon (IFN)γ (reviewed by Gao et al. [2009]).
The extracellular matrix
Fibrosis accumulation is a dynamic process resulting from a wound-healing response to acute or chronic liver injury [Albanis and Friedman, 2001]. While acute injury triggers scar formation that resolves, in chronic injury repeated and overlapping phases of inflammation disrupt homeostasis, and scar tissue progressively replaces the liver parenchyma.
The formation of an interstitial scar-type, collagen-rich matrix represents a change in both the quality and quantity of the extracellular matrix (ECM). There is overproduction and deposition of fibrotic proteins such as fibrillar collagens [Desmouliere et al. 1997; Friedman, 2004b], of which type I predominates, in addition to collagens III and IV [Rojkind et al. 1979]. There is also accumulation of the noncollagen matrix proteins that includes fibronectin, undulin, elastin, laminin, hyaluronan, and proteoglycans [Benyon and Iredale, 2000]. The expression and release of some of those proteins into circulating blood has been used to assess liver fibrosis in several noninvasive methods (see below).
In order to preserve matrix homeostasis, the ECM also contains MMPs. MMP-1, −8, and −13 degrade the fibrillar collagen types I and III that predominate in fibrosis, while MMP-2 and −9 degrade collagen IV as well as denatured fibrillar collagens. HSCs are the key source of both MMPs in liver. Although the increase in MMP production should control the excessive increase in the ECM, it can also promote injury. Early increases in MMP, particularly MMP-2, degrade normal matrix and recruit cells that can amplify fibrosis [Arthur, 2000; Takahara et al. 1997]. In addition, there is also enhanced secretion of tissue inhibitor of metalloproteinases-1 and −2 (TIMPs) by HSCs during progressive tissue injury and cellular activation (Figure 3). TIMPs are key regulators of MMPs, by blocking their collagenolytic activity. In addition, TIMP1 is anti-apoptotic towards HSCs, in part through induction of Bcl-2, thus promoting the survival of fibrogenic cells [Yoshiji et al. 2000]. In human liver the degree of TIMP-1 expression correlates with extent of fibrosis [Benyon et al. 1996].
Fibrosis versus resolution. As hepatic stellate cells (HSCs) activate and transdifferentiate to myofibroblasts, gene expression is altered, for example loss of peroxisome proliferator-activated receptor gamma (PPARγ) expression. Perpetuation involves distinct changes in HSC behavior that promote proliferation and contractility. In addition, paracrine stimulation, especially by platelet-derived growth factor (PDGF), increases the recruitment of myofibroblasts. Paracrine and endocrine signaling drive extracellular matrix (ECM) synthesis. Reducing the cause of injury may promote clearance of activated HSCs by apoptosis, senescence or reversion to quiescence. In addition, matrix degradation is augmented by increasing matrix metalloproteinase (MMP) activity and decreasing tissue inhibitor of metalloproteinase (TIMP). Illustration courtesy of Alessandro Baliani, Copyright © 2011.
The consequence of an imbalance between production of collagen and its degradation results in a net deposition of fibrillar collagen. Next, the collagen undergoes crosslinking mediated by transgluraminases and lysyl oxidases, which stabilize the ECM [Barry-Hamilton et al. 2010; Grenard et al. 2001] and further increase its resistance to degradation [Issa et al. 2004]. As disease progresses, the collagen bundles form bridges that later may provoke the development of regenerative nodules and architectural distortion, with impairment of liver function typical of liver cirrhosis (Figure 3).
From fibrosis to cirrhosis
The progression from fibrosis to cirrhosis occurs over decades, with an average of 20–30 years from the time of infection to cirrhosis in chronic HCV, the most common cause of cirrhosis in the US. The ‘injury–cirrhosis’ interval may vary; in some patients the process can take 40 years (‘slow fibrosers’) while in others it can be shorter than 15 years (‘rapid fibrosers’) [Sobesky et al. 1999], despite the same etiology. This realization is particularly important in the setting of HCV, because antiviral therapies to date have had suboptimal response rates and high adverse events, so the decision whether to treat must take into account the likelihood of fibrosis progression.
While viral factors (i.e. genotype or viral load) do not influence fibrosis progression in patients with HCV, host factors play an important role [Poynard et al. 1997]. Numerous association studies have investigated the role of gene polymorphisms in the progression of liver fibrosis and/or development of cirrhosis in patients with chronic liver diseases. Genetic variations are involved in either the susceptibility to progression of liver disease, persistence of HCV infection, and/or the response to antiviral therapy.
In a series of patients with chronic HCV, polymorphisms in the angiotensinogen gene, the angiotensin II precursor, as well as the TGF-β1 genes, were uncovered as determinants of fibrosis progression [Powell et al. 2000]. Patients with single nucleotide polymorphisms (SNPs) in both genes progress more rapidly than those having only one SNP. Recently, seven predictive SNPs were combined into a cirrhosis risk score (CRS), which can be used to determine a patient’s heritable risk of fibrosis progression when they have chronic HCV infection [Huang et al. 2007]. Several HLA-II alleles and polymorphisms of genes involved in the immune response (e.g. mannose binding lectin and haptoglobin) also influence the susceptibility or resistance to persistent HCV infection [Sasaki et al. 2000; Louagie et al. 1996]. Similarly, variations in IL-10, and MxA, an interferon-induced protein, can predict an individual’s response to IFN therapy [Yee et al. 2001; Hijikata et al. 2000].
Additional correlations between SNPs and specific disease risk have been described, but have not yet begun to impact on routine clinical practice (see Bataller et al. [2003a] for a review).
Because standard clinical indices cannot distinguish between minimal and advanced fibrosis, the ability to define clinical and genetic risk factors will become a valuable tool to predict rapid fibrosis progression and perhaps the risk of liver failure. This kind of information will eventually define the need for therapy, as stated in the National Institutes of Health (NIH) consensus statement, ‘treatment is recommended for patients with an increased risk of developing cirrhosis’ [National Institutes of Health, 2002]. Improved genetic information will hopefully lead to a therapy response-based gene signature. This may allow tailoring of individualized therapy with better efficacy and fewer side effects.
Assessment of liver fibrosis
A major difficulty in developing antifibrotic therapies is the lack of accurate and established techniques to estimate fibrosis regression in response to therapy. Liver biopsy has long been the gold standard for assessing staging, but has several limitations. First, biopsy is an invasive technique which has associated morbidity; pain occurs in 20% of patients and major complications (such as bleeding or hemobilia) in 0.5% [Cadranel et al. 2000]. The bleeding rate (0.5%) has not changed significantly in recent years, according to a large multicenter study from the HALT C trial sponsored by NIDDK-NIH [Seeff et al. 2010]. The primary factor that appeared to contribute to bleeding risk was platelet count rather qualitative factors such as operator experience, needle size or the use of ultrasound to localize the site. Second, the small size of the biopsy makes it prone to sampling variability [Bedossa et al. 2003]. Third, the interpretation of the histologic changes can be problematic with inter- and intra-observer variation [Regev et al. 2002]. On the other hand, at least some correlation between biopsy stage and outcomes has begun to emerge. In the NIH-HALT C cohort, a correlation was found between the Ishak fibrosis stage and clinical outcomes, the need for liver transplantation, and liver-related deaths in patients with chronic HCV. However, even in this study up to 25% of the biopsy samples were fragmented, which significantly diminished the ability to draw correlations between biopsy findings and clinical outcomes [Everhart et al. 2010].
The current focus in the field is to develop noninvasive techniques to assess fibrosis stage. Ongoing efforts include serum markers and imaging based on ultrasound, CT, and MRI. The goal is to develop tests with high specificity and sensitivity to estimate fibrosis and predict outcomes.
Class II and class I serum biomarkers
Class II biomarkers are indirect methods using serum biochemical and/or hematological tests, and are based on the detection of common functional alterations in the liver. In contrast, class I biomarkers are associated with the process of fibrogenesis, and their presence in the serum is the result of the increased turnover of ECM. The clinical acceptance of these biomarkers is still low in the United States. Simple and readily available biomarkers have low accuracy in predicting liver fibrosis and more advanced markers have an unacceptable cost–benefit ratio. Thus, liver biopsy still remains the gold standard for diagnosis of fibrosis. Additional noninvasive alternatives are being developed, and their implementation will be essential to test antifibrotic drugs in clinical trials [Jarcuska et al. 2010].
Measures using combinations markers have comparable ranges of accuracies as the individual markers. The best-validated methods are Fibrotest (gamma-glutamyl transpeptidase [GGT], haptoglobin, bilirubin, apolipoprotein A1, alpha-2-macroglobulin) and enhanced liver fibrosis test (ELF) (N-terminal propeptide of collagen type III, hyaluronic acid, TIMP-1, age). Recent data indicate that the results from these serum tests may be more accurate than biopsy findings in predicting risk of decompensation, survival without HCV-related complications and overall survival [Mayo et al. 2008; Ngo et al. 2006].
Imaging techniques
Major advances in imaging technologies enable better detection of fibrosis. Transient elastography (FibroScan) is the most widely used noninvasive method for assessing the degree of liver fibrosis in Europe [Sandrin et al. 2003]. The technology is based on pulse-echo ultrasound to obtain liver stiffness measurements (LSM). It is rapid, noninvasive, reproducible, and acquires information from a much larger portion of the tissue than liver biopsy; therefore, the risk of sampling error is significantly lower. Transient elastography best distinguishes between stages at either end of the fibrosis spectrum. Its utility is limited, however, in patients with narrow intercostal spaces or morbid obesity. The stiffness may also be elevated in patients with acute hepatitis, probably reflecting edema and inflammation rather than fibrosis [Arena et al. 2008]. Using the combination of clinical variables of donor age and bilirubin with LSM has proven to be an effective strategy to estimate fibrosis progression rate, and to distinguish between rapid and slow fibrosers after liver transplant patients with recurrent HCV [Carrion et al. 2010]. Most recently, liver stiffness values as well as FibroTest have been more closely correlated with risk of decompensation over 5 years than liver biopsy [Vergniol et al. 2011] Recently a combination of FibroMeter and Fibroscan has provided a new classification for the noninvasive staging of liver fibrosis in patients with chronic HCV [Boursier et al. 2011]. This is similar to the Lok score in which liver stiffness is combined into a noninvasive algorithm for the assessing the likelihood of esophageal varices in cirrhotic patients [Stefanescu et al. 2011].
Acoustic radiation force impulse (ARFI) imaging technology has been implemented as a method to assess liver fibrosis. Recent studies have shown excellent diagnostic accuracy in identifying significant fibrosis and cirrhosis in patients with various liver diseases [Friedrich-Rust et al. 2009]. The technology can be adapted to existing ultrasound devices, making it potentially more accessible than transient elastography.
New MRI-based technologies are also gaining interest: contrast-enhanced MRI, diffusion-weighted MRI, and magnetic resonance elastography [Talwalkar et al. 2008]. The advantages are their assessment of the entire liver parenchyma, lack of an acoustical window requirement, and operator independence. The disadvantages are their cost and time-consuming nature.
Resolution of fibrosis
Exciting clinical evidence has demonstrated that cirrhosis not only undergoes histological reversion [Friedman and Bansal, 2006], but can also be associated with improved clinical outcomes [Mallet et al. 2008]. Resolution of fibrosis is likely due to increased activity of interstitial collagenases and decreased TIMP expression, contributing to the clearance of activated HSCs. In turn, clearance of activated HSCs can result from apoptosis, senescence and/or reversion to quiescence (Figure 3). These pathways are supported by findings in patients with chronic HCV in whom fibrosis progression was associated with progressive loss of HSC apoptosis [Gonzalez et al. 2009].
More recently, cellular senescence of HSCs during resolution has been documented [Krizhanovsky et al. 2008]. Cellular senescence is a genetically controlled program preventing cell division once cells exceed a finite proliferative capacity. The genetic profile of senescent HSCs is defined in part by induction of p53, p21, and p16. Furthermore, the cells are more susceptible to attack by NK cells [Krizhanovsky et al. 2008] in vitro and in vivo, thereby additionally facilitating fibrosis resolution.
Reversion of activated HSCs to quiescent cells may be an additional pathway that contributes to HSC clearance. This event is suggested by the re-expression of genes associated with a quiescent phenotype, such as peroxisome proliferator-activated receptor gamma (PPARγ) or its ligands [Hazra et al. 2004].
Antifibrotic therapy
The increasing evidence that fibrosis is a dynamic and reversible process, the clarification of the underlying sources and mediators of fibrosis progression, and advances in noninvasively assessing fibrosis have generated enthusiasm towards developing effective antifibrotic drugs. To date, however, no drug has been approved as an antifibrotic. In reality, there may already be many existing drugs with well-established safety profiles, whose mechanism of action will be also antifibrotic even though they have been developed for other indications. Challenges remain, as noted, including the typically decades-long natural history of disease that will require long-term pharmacologic intervention to prevent or reverse cirrhosis, and the lack of a standardized, accepted noninvasive endpoints for fibrosis assessment.
Ongoing antifibrotic clinical trials Registered at ClinicalTrials.gov.

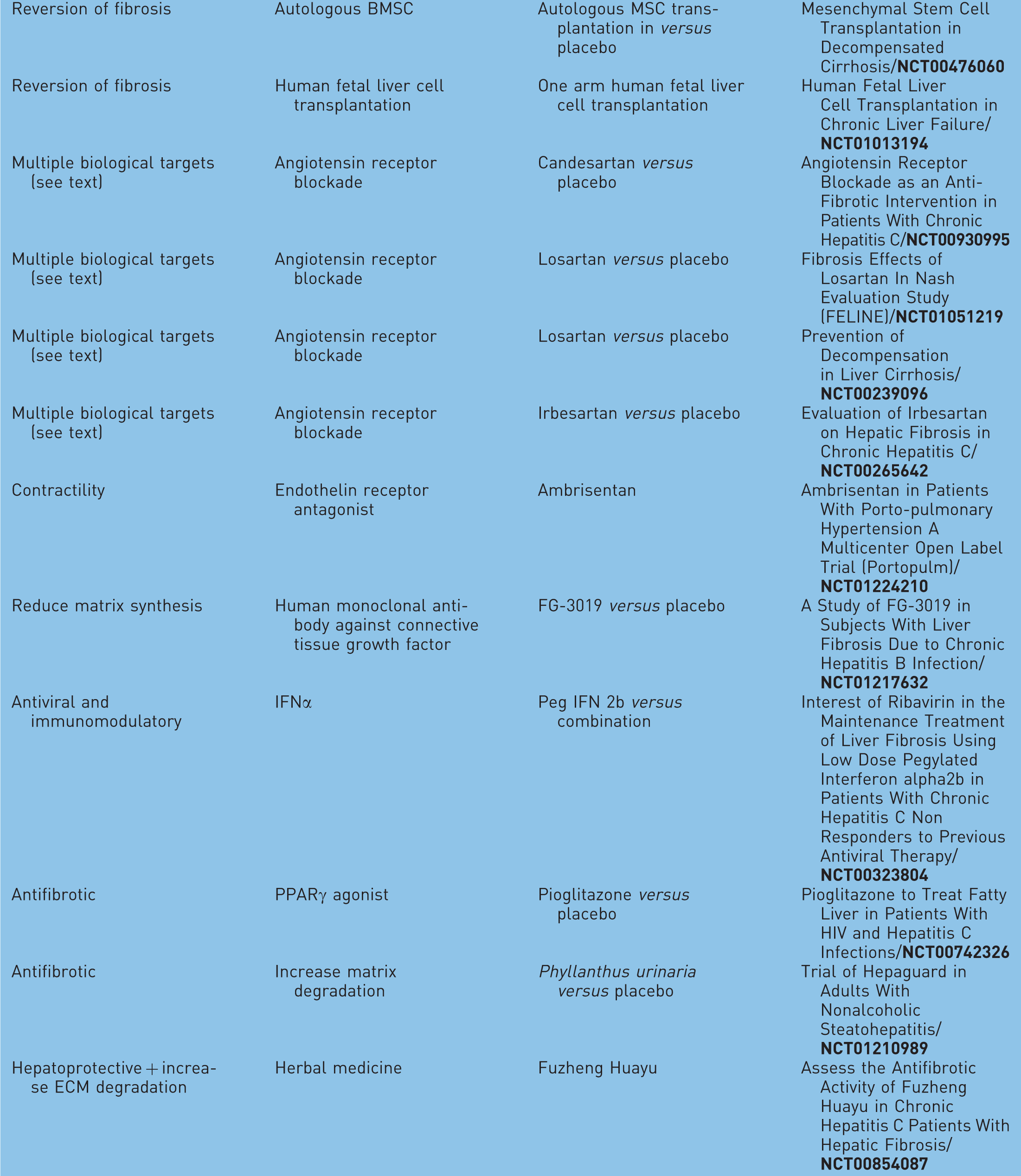
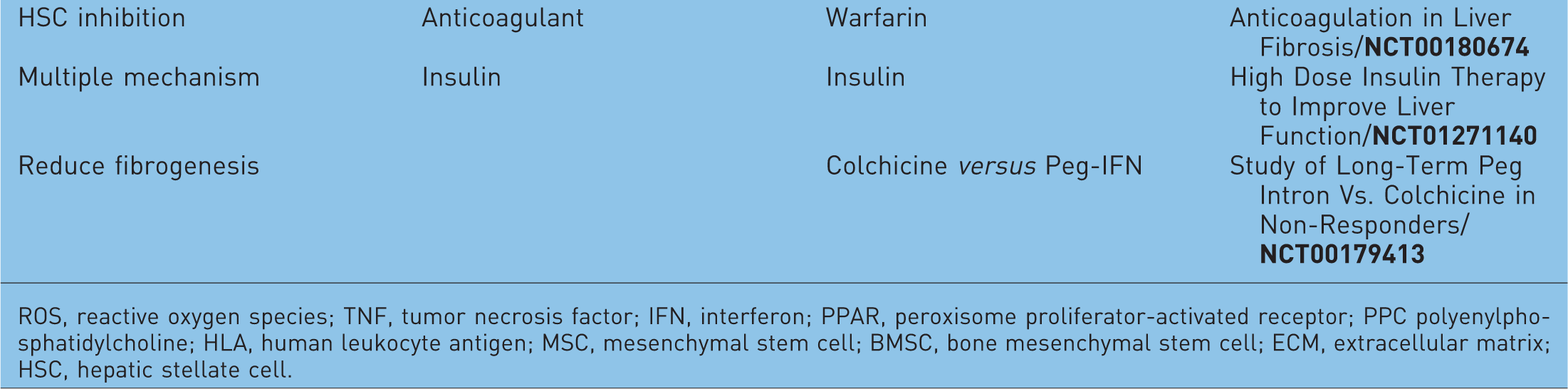
ROS, reactive oxygen species; TNF, tumor necrosis factor; IFN, interferon; PPAR, peroxisome proliferator-activated receptor; PPC polyenylphosphatidylcholine; HLA, human leukocyte antigen; MSC, mesenchymal stem cell; BMSC, bone mesenchymal stem cell; ECM, extracellular matrix; HSC, hepatic stellate cell.
Completed trials of antifibrotic agents in patients with liver disease.

HCV, hepatitis C virus; TNF, tumor necrosis factor; IL, interleukin; NASH, nonalcoholic steatohepatitis.
There are several points of attack in developing antifibrotic agents that are described in detail in the following sections:
Eliminate the cause(s) of injury and their mediators. Reduce inflammation and the immune response. Target specific signaling: receptor–ligand interaction, intracellular signaling. Reduce fibrogenesis, inhibit matrix synthesis. Resolve fibrosis by:
○ increasing scar matrix degradation; ○ stimulating apoptosis of stellate cells; ○ BM or cell transplantation. Miscellaneous.
Eliminate the cause(s) of injury and their mediators
Liver injury provokes scar formation that is driven by different mediators depending on the etiology [Bataller and Brenner, 2005]. To date, clearance or control of the underlying etiology is the most effective antifibrotic treatment. In chronic HCV the cleaved viral proteins are direct mediators of hepatocyte injury. Bound to the MHC-I/MHC-II, they may lead to oxidative stress and pro-inflammatory response. Direct HSC activation by viral proteases has also reported [Bataller et al. 2004]. Yet, patients who respond to therapy and have cleared the virus can effectively abrogate the injury signal, which can lead to reversion of fibrosis even with advanced fibrosis [Abergel et al. 2004]. In contrast, if the infection (i.e. trigger) persists, no therapy can yet attenuate fibrosis. Most recently a large multicenter 5-year trial of low-dose IFN monotherapy showed no benefit on fibrosis. [Haque and Yoshida, 2009] (Table 2). Fibrosis and cirrhosis can be reversible in patients who have autoimmune hepatitis that respond to steroids [Dufour et al. 1997], as well as following removal of excess iron in hemochromatosis or copper in Wilson disease. Similarly, abstinence in alcoholic liver disease, decompression in biliary obstruction [Hammel et al. 2001], and weight loss in patients with NASH [Dixon et al. 2004] all can achieve remarkable fibrosis regression. Therapeutic approaches such as Roux-en-Y gastric bypass can reduce both steatosis and fibrosis in NASH patients [Furuya et al. 2007]. The use of the PPARγ agonist, rosiglitazone, attenuated steatosis and fibrosis in NASH patients in one study [Neuschwander-Tetri et al. 2003]. However, these effects were not seen in a more recent multicenter NIH-sponsored trial [Sanyal et al. 2010].
Reduction of bile acid-induced membrane injury may underlie the response of some cholestatic diseases to ursodeoxycholic acid, a nontoxic bile acid, that binds to cell membranes and is presumably cytoprotective [Rockey, 2008]. This effect is thought to reduce inflammation and downstream fibrogenesis [Nava-Ocampo et al. 1997]. Ursodeoxycholic acid may impede progression of fibrosis in primary biliary cirrhosis (PBC) through effects on biliary ductal inflammation, particularly if the drug is initiated early in the disease course. However, extensive human studies in a variety of liver diseases have failed to establish a direct antifibrotic effect of ursodeoxycholic acid.
As hepatocytes are injured, they release apoptotic bodies, cytokines, and inflammatory signals (Figure 2). Therefore, reducing the mediators or inhibiting apoptosis by anti-apoptotic drugs (e.g. caspase 3 inhibitor) is a rational approach, but unfortunately has not been successful in clinical trials. All trials using caspase inhibitors terminated because of significant laboratory abnormalities and adverse events among some clinical trial participants (ClinicalTrials.gov).
Reduce inflammation and the immune response
Since persistent inflammation almost always precedes and accompanies fibrosis, drugs that target the inflammatory cascade typically have antifibrotic activity. Moreover, because the vicious cycle of scar formation is initiated by oxidative stress, targeting ROS generation also reduces the inflammatory response, which will attenuate HSC activation and fibrogenesis.
Antioxidants can attenuate ROS effects and hold promise as potential antifibrotic therapies, provided that sufficient antioxidant activity can be delivered to the sites of injury within the liver. Antioxidants exert a preventive effect on hepatocyte injury but may also be directly antifibrogenic. Antioxidant compounds include food supplements and drugs that have tested in animal models and human trials, including S-adenosyl-L-methionine, silymarin, phosphatidylcholine, N-acetylcysteine, resveratrol [Kawada et al. 1998], and vitamin E [Sanyal et al. 2010].
S-adenosyl-L-methionine (SAMe) reduces ROSs and inhibits HSC activation in a model of alcoholic liver injury [Karaa et al. 2008] and in a clinical trial [Martinez-Chantar et al. 2002]. In a long-term randomized, placebo-controlled, double-blind, multicenter clinical trial of SAMe in patients with alcoholic liver cirrhosis, SAMe improved survival or delayed liver transplantation [Martinez-Chantar et al. 2002]. S-nitroso-N-acetylcysteine (SNAC), a nitric oxide donor, has an antifibrotic effect in rats with secondary biliary cirrhosis [Vercelino et al. 2010] by down-regulating expression of genes and modulating intracellular signaling pathways; this activity is independent of its antioxidant capacity [Martinez-Chantar et al. 2002]. The agent has not been validated in human trials yet, however.
Recent results have suggested that Vitamin E is effective in patients with NASH [Harrison et al. 2003]. In a large NIH trial (‘PIVENS’ trial, see Table 2) in 247 patients treated for 96 months, vitamin E led to clear histological regression, with no fibrosis progression [Sanyal et al. 2010].
NADPH oxidase, an enzyme that generates oxidant stress, is activated by angiotensin II (Ang II) in inflammatory regions within the liver [De Minicis and Brenner, 2007]. Long-term administration of losartan (an Ang II inhibitor) decreases the expression of NADPH oxidase, and the fibrogenic genes collagen I, MMP2 and urokinase [Colmenero et al. 2009].
Suppression of ROS and NADPH oxidase 4, which are induced by TGF-β1, are the therapeutic targets of polyenylphosphatidylcholine in suppressing HSC activation. However, a human trial in alcoholic liver disease showed limited efficacy [Lieber et al. 2003].
Because inflammation promotes the progression to liver fibrosis, the use of anti-inflammatory drugs has a clear rationale. Corticosteroids are still used as first-line therapy in autoimmune hepatitis [Ishibashi et al. 2007]. However, the suppression of fibrogenesis is sometimes incomplete and fibrosis may develop eventually despite steroids in many patients [Albanis and Friedman, 2001]. In experimental models, the use of dexamethasone in cell culture has reduced TGF-β signaling in primary HSCs [Bolkenius et al. 2004]. However, targeting of dexamethasone to Kupffer cells has also accelerated fibrogenesis by increasing TIMP-1 in rats with bile duct ligation [Melgert et al. 2001]. These dual responses emphasize the complexity of fibrosis signaling and the difficulty of using a drug like a corticosteroid, which has multiple targets of action.
Another anti-inflammatory strategy is to neutralize inflammatory cytokines using specific cytokine and/or receptor antagonists. In animals, anti-TNFα effectively reduces the serum elevation in liver enzymes and inflammatory cytokines including IL-6 [Li and Friedman, 1999] and TGF-β1 [Bahcecioglu et al. 2008]. The reduction of these cytokine levels are accompanied by diminished necrosis and inflammation in tissue sections. Because TNFα is upregulated in alcoholic liver disease, several clinical trials have tested different anti-TNF compounds (e.g. infliximab, etanercept, and pentoxifilline) to reduce complications and improve outcomes, rather than to decrease fibrosis. However, etanercept, a TNF soluble receptor administered to patients with moderate to severe alcoholic hepatitis, had no impact on mortality at one month, and treated patients had a greater mortality after 6 months, which was attributed to an increased risk of infections (Table 2). Histologic data were not evaluated [Boetticher et al. 2008]. In contrast, pentoxyphylline reduced short-term mortality in one study, but no antifibrotic activity was noted [Akriviadis et al. 2000]. Recently, a double blind, placebo-controlled study in patients with advanced cirrhosis (Child Pugh class C) compared pentoxyphylline with placebo (Table 2). The drug did not decrease short-term mortality, however it reduced the risk of complications (e.g. bacterial infection, renal insufficiency, hepatic encephalopathy, or gastrointestinal hemorrhage) and increased survival in the subgroup of patients without complications [Lebrec et al. 2010]. Although fibrosis scores were not evaluated in this study, there was a clear trend in changing the disease course, although it is unclear whether the drug’s effect is through reduced TNFα activity or signaling, or through another pathway.
Targeting receptor–ligand interactions and intracellular signaling
The discovery of membrane and nuclear receptors expressed by HSCs that have been previously identified in other tissues has opened new possibilities for antifibrotic therapies. These targets include the renin-angiotensin system, and receptors for serotonin, cannabinoids, and endothelin 1 (ET-1), as well as intracellular signaling involving the nuclear receptors PPARγ, farnesoid X receptor (FXR), pregnane X receptor (PXR), and liver X receptor (LXR). NF-κB is also a critical signal controlling HSC survival and cellular responses.
Neurochemical receptors
At least three classes of neurotransmitters are expressed in myofibroblasts, cannabinoids, opioids, and serotonin (5HT), and each regulates key fibrogenic activities in the cell.
Cannabinoids are a very attractive target for modulating hepatic fibrosis. Two G protein coupled receptors, CB1 and CB2, are expressed in liver. The CB1 receptor is upregulated in myofibroblasts and promotes fibrosis. Indeed, daily cannabis (marijuana) increases fibrosis progression in chronic HCV, since it primarily signals through CB1 [Hezode et al. 2005]. Conversely, CB2 receptor signaling is antifibrotic [Julien et al. 2005], but may amplify inflammation [Deveaux et al. 2009; Munoz-Luque et al. 2008]. As a result of these findings, CB1 antagonism is a more promising strategy. In a 2-year randomized, placebo-controlled human trial, the CB1 antagonist Rimonabant led to significant weight loss and improved metabolic function, decreased triglycerides and improved insulin resistance. However, the drug was withdrawn because of a high percentage of patients who developed clinical depression, a predictable consequence of CB1 receptor antagonism in the central nervous system (CNS) [Gelfand and Cannon, 2006]. As a result, current efforts are directed towards CB1 antagonists that do not cross the blood–brain barrier. Two such ongoing trials are listed at ClinicalTrials.gov for metabolic syndrome, but an antifibrotic endpoint is not described in their study designs.
Opioids and their receptors are other potential therapeutic targets, and their use has been ongoing in patients with primary sclerosing cholangitis (PSC). Endogenous opioids have proliferative and profibrogenic activity in HSCs [De Minicis et al. 2008]. Naltrexone, an opioid antagonists used as an antipruritic drug in PBC, has antifibrotic activity in animal models of liver disease [Ebrahimkhani et al. 2006].
Human myofibroblasts also express 5HT receptor subtypes, which mediate proliferation and fibrogenesis. Antagonism with methiothepin or spiperone increases myofibroblast apoptosis and reduces fibrosis in an animal model [Ruddell et al. 2006].
The renin–angiotensin system
Angiotensin II (Ang II) is a key player in fibrogenesis. Ang II is secreted by HSCs, and binds to the AT1 receptor, also expressed by HSCs [Bataller et al. 2003b]. Ang ІІ induces contraction and proliferation of human HSCs [Bataller et al. 2000], as well as increasing collagen I gene expression in vitro [Tharaux et al. 2000]. AT1a receptor knockout mice have reduced lipid peroxidation products, inflammation and fibrosis following bile duct legation [Yang et al. 2005]. Consequently, blocking the renin–angiotensin system (RAS) by ACE inhibitors or AT1 receptor blockers (ARBs) may be an effective strategy in the treatment of liver fibrosis, and is already undergoing testing in human trials. Long-term administration of losartan to patients with chronic HCV leads to decreased NADPH oxidase, decreased inflammation, and reduced fibrogenesis. Paired biopsies have shown a decrease of at least one stage of fibrosis in half of the treated patients, compared with only 5–24% of patients with spontaneous regression [Ghany et al. 2003]. Evaluation of collagen accumulation supported the same trend with a mean increase of only 1% after 18 months of therapy compared with progression in 50% of control patients [Colmenero et al. 2009]. The strength of these data is undermined by a lack of randomization. An association with hypertension, possibly via the RAS, and a beneficial role of Ang II blockade in HCV-related fibrosis has been reported from a randomized control trial comparing three groups; HCV patients with hypertension treated with ARB compared with non-ARB treatment, and compared with nonhypertensive patients [Corey et al. 2009]. Administration of an ARB, compared with other antihypertensive drugs in patients with recurrent HCV after liver transplantation, was associated with less progression in inflammation but not in fibrosis [Cholongitas et al. 2010]. Recently, in the HALT-C cohort ACEi/ARB therapy did not retard the progression of hepatic fibrosis [Abu Dayyeh et al. 2011]. Further controlled studies are needed to evaluate the effect of long-term efficacy of ARBs.
Endothelin 1
ET-1 and nitric oxide (NO) are major counter-regulators controlling HSC contractility (Figure 3). ET-1 is vasoconstrictor with a potent effect on the hepatic vasculature. High levels of ET-1 and endothelin receptors are present in cirrhosis [Yokomori et al. 2001]. The blockade of ET-1 type A receptor and the administration of vasodilators (prostaglandins E2 and NO donor) exert an antifibrotic activity in rodents and also improve portal hypertension [Cho et al. 2000]. NO is a highly reactive free radical that has diverse physiological and pathological roles. There is increased NO and activity of inducible NO synthase (iNOS) in hepatic injury (e.g. ischemic-reperfusion injury or cirrhosis). Blocking iNOS by oral administration of the inhibitor RF260330 in a rat model reduced fibrosis by inhibiting cytokine production (e.g. TGF-β1) [Kikuchi et al. 2007]. There are no human data evaluating the drug (see Table 1).
Adipokines
As obesity becomes increasingly prevalent, new findings are emerging that define the role of adipose derived hormones, or adipokines, in contributing to the complications of obesity, including the metabolic syndrome and hepatic fibrosis.
Metabolic abnormalities are considered the ‘first hit’ of liver injury in obesity-related liver disease, followed by oxidative stress and inflammation [Marra et al. 2005]. Leptin and its natural antagonist adiponectin are key adipokines secreted by adipose tissue and stromal cells, especially HSCs [Ikejima et al. 2007]. Blood leptin levels correlate with fat mass, and as obesity progresses, elevated leptin levels signal through their specific receptors to promote fibrogenesis by JAK/STAT signaling [Choi et al. 2010]. In contrast, adiponectin levels are inversely correlated with body fat and antagonize fibrogenesis [Wedemeyer et al. 2009].
In addition, ghrelin, a gut hormone that plays a major role in the regulation of food intake, is decreased in advanced liver disease and correlates with fibrogenic gene expression. A polymorphism of the ghrelin gene reportedly influences the progression of fibrosis in patients with chronic HCV [Moreno et al. 2010]. In rats, recombinant ghrelin exerts hepatoprotective and antifibrotic effects in liver [Moreno et al. 2010]; further studies are needed to determine its safety, tolerability, and efficacy in human liver disease.
Tyrosine kinase receptors
Many proliferative cytokines, including PDGF, fibroblast growth factor (FGF), and TGF-α signal through tyrosine kinase receptors, a family of cell surface molecules that phosphorylate specific tyrosine residues following binding of ligand. Antagonism of pathways that mediate PDGF or VEGF signals reduces HSC proliferation [Gonzalo et al. 2007; Tugues et al. 2007]. For example, sorafenib, a multiple receptor tyrosine kinase inhibitor approved for therapy in hepatocellular carcinoma, targets the PDGF receptor and Raf/ERK signaling pathways [Wang et al. 2010]. Similarly, gleevec, a small molecule tyrosine kinase antagonist used in chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors, is antifibrotic [Yoshiji et al. 2005]. Moreover, the combination of gleevec with an ACE inhibitor could be especially potent and safe [Yoshiji et al. 2006]. Nilotinib is another novel small molecule tyrosine kinase inhibitor of Bcr-Abl. Nitolinib reduces liver injury and fibrosis through multiple mechanisms [Liu et al. 2011]. Nilotinib significantly inhibits PDGF and TGF-β-simulated phosphorylation of ERK and Akt, indicating that it represents a potentially effective antifibrotic agent [Liu et al. 2011].
Nuclear receptors
HSCs express a diverse group of nuclear transcription factor receptors, including PPARγ, FXR, and PXR [Mann and Mann, 2009]. PPARγ is strongly expressed in quiescent HSCs and falls rapidly when they activate [Marra et al. 2000]. Thiazolidones, antidiabetic agents that are PPARγ agonists, reduce collagen expression and HSC activation in culture [Galli et al. 2002]; however, clinical trials have been unsuccessful to date [McHutchison et al. 2010]. Although there was no evidence of reduced fibrosis either in HCV patients [McHutchison et al. 2010], or in NASH patients, there was a trend towards improvement compared with placebo (p = 0.04), but a significant improvement in steatosis, inflammation and insulin resistance [Sanyal et al. 2010].
Binding of bile acids by FXR is antifibrotic, by reducing collagen I expression [Fiorucci et al. 2004]. Furthermore, this signaling cascade inhibits TIMP-1 expression, which reduces the survival of activated HSC [Fiorucci et al. 2005]. INT-747 is a synthetic bile acid that is currently in clinical trials for cholestatic liver diseases (Table 1); its effects could also be mediated via PXR since PXR is activated by a range of exogenous and endogenous ligands (e.g. rifampicin, steroids, bile acid) that reduce expression of TGF-β and inhibit proliferation [Wallace et al. 2008].
LXRs function as key regulators of lipogenesis and modulate the immune system [Castrillo and Tontonoz, 2004; Schultz et al. 2000]. Their role in fibrosis is still under investigation. Recently, Beaven and colleagues demonstrated that LXR signaling modulates suppression of HSC activation and inflammatory genes (MCP, IL-6). In addition, LXR-deficient mice have increased secretion of inflammatory mediators and enhanced susceptibility to liver fibrosis in different models [Beaven et al. 2011]. To date, the use of LXR agonists as a potential antifibrotic agent has been disappointing because of hepatotoxicity due to induction of de novo lipogenesis [Schultz et al. 2000].
Inhibiting fibrogenesis
Inhibition of matrix production is one of the most appealing therapeutic strategies and the list of target cytokines involved in this process is growing. TGF-β1 is the most potent inducer of collagen I and other matrix constituents, and thus inhibiting its actions remain a major focus of antifibrotic efforts in liver. Signals downstream of the TGF-β receptors include the Smads, upon which many intra and extracellular signals converge to fine-tune TGF-β’s effects. In addition, TGF-β1 stimulates collagen in HSCs through a C/EBPβ-dependent mechanism [Garcia-Trevijano et al. 1999].
Disruption of TGF-β synthesis or activity will reduce collagen synthesis and scar formation and accelerate matrix degradation. Efforts to antagonize TGF-β (e.g. soluble TGF-β receptor) [George et al. 1999] or decrease its proteolytic activation (e.g. Camostat mesilate) [Okuno et al. 2001] have been evaluated in experimental models. Connective tissue growth factor (CTGF/CCN2) is also a potent fibrogenic signal towards HSCs, and is upregulated by hyperglycemia and hyperinsulinemia [Paradis et al. 2001]. FG-3019, a human monoclonal antibody against CTGF, has been tested in lung fibrosis and its now being assessed as a treatment for liver fibrosis in patients with HBV in Asia (Table 1).
Recently, Oltipraz [5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione], a drug previously studied as a cancer chemopreventive agent, was found to inactivate HSCs and suppress TGF-β1 in animal models [Kang et al. 2002]. Early studies demonstrated efficacy in enhancing regeneration of cirrhotic liver, reducing fibrotic and cirrhotic nodules, and eliminating accumulated extracellular matrix in animal models. However, a double-blind, placebo-controlled trial failed to show efficacy. A correlation was noted between plasma TGF-β1 and the Ishak fibrosis score, suggesting that circulating TGF-β1 serves as a possible indicator of the need for fibrosis treatment [Kim, 2011].
Environment, deitery and behavioral risk factors also contribute to fibrosis progression. Coffee and caffeine consumption are associated with reduced hepatic fibrosis when at least 2.25 cups of coffee equivalents are consumed daily [Modi et al. 2010; Freedman et al. 2009; Ruhl and Everhart, 2005]. The antifibrotic effect may be due to antagonism of hepatic adenosine A2A receptor activity, which in animal models reduces the fibrogenic activity of HSCs [Chan et al. 2006]. These findings suggest that selective adenosine receptor antagonists with more favorable pharmacokinetics might offer even greater protection from the development of hepatic fibrosis and cirrhosis [Cronstein, 2010]. In addition, adenosine generated by ethanol metabolism plays an important role in ethanol-induced hepatic steatosis via both A1 and A2B receptors, suggesting that targeting adenosine receptors may be effective in the prevention of alcohol-induced fatty liver [Robson and Schuppan, 2010]. In a cohort study, the relative risk of alcoholic cirrhosis (199 subjects) for coffee drinking (versus none) was reduced inversely with the amount consumed from 0.2 to 4.0 or more cups per day (95% confidence interval [CI] 0.1–0.4; p < 0.001) [Klatsky et al. 2006]. Concurrently, caffeine has been found to inhibit TGF-β-stimulated CTGF expression in hepatocytes via PPARγ and SMAD2/3-dependent pathways [Gressner et al. 2008].
Direct inhibition of collagen synthesis in response to a range of drugs and herbal compounds is another potential strategy. Pirfenidone (5-methyl-1-phenyl-2-(1H)-pyri-) is a small orally bio-available molecule that appears to inhibit collagen synthesis. Efficacy has been evaluated in idiopathic pulmonary fibrosis and in 15 patients with HCV-related fibrogenesis. In the liver trial, fibrosis was reduced in 5 of 15 patients (30%) following 12 months of treatment [Rockey, 2008].
Colchicine is a plant alkaloid that is classified as anti-inflammatory drug. Its main mechanism of action is by inhibiting polymerization of microtubules, thereby interfering with collagen secretion. In clinical trials of patients with PBC treated with colchicine, mortality and transplantation rates were unaffected, but laboratory values [Bodenheimer et al. 1988] and fibrosis markers improved (e.g. PIIINP) [Nikolaidis et al. 2006]. In a long-term controlled study, colchicine prolonged survival in patients with mild to moderate cirrhosis regardless of the cause [Kershenobich et al. 1988]. This study was criticized, however, because of the high nonliver mortality in the control group and other methodological concerns. In a separate meta-analysis including 1138 subjects, colchicine had no effect on fibrosis or mortality [Rambaldi and Gluud, 2001]. Overall, colchicine does not appear to reduce hepatic fibrosis, and it cannot therefore be recommended as a primary antifibrotic treatment.
Herbal medicines are broadly used in Asia and may merit further development. The most popular is sho-saiko-to, which reduces synthesis of hepatic type I and III collagen and hydroxyproline content [Sakaida et al. 1998]. Similarly Salvia genus is considered antifibrotic as well. Curcumin has also been studied extensively, as its activities include inhibition of ECM formation, suppression of proliferation and increased apoptosis in HSCs [O'Connell and Rushworth, 2008]. Although there is compelling efficacy based on animal models, rigorous data from human trails are lacking, but clinical trials are ongoing (Table 1).
Resolution of fibrosis
Increased matrix degradation
Enhancing the degradation of the collagen-rich ECM (fibrotic scar) is another approach to developing antifibrotic drugs. Possible strategies are to increase the activity of endogenous matrix-degrading enzymes or to administer degrading enzymes using gene therapy. MMP-8 and urokinase-type plasminogen activator stimulate collagen degradation in vivo [Siller-Lopez et al. 2004; Salgado et al. 2000]. Upregulation of MMPs, (e.g., MMP-8, MMP-1) using adenovirus transfection leads to attenuated fibrosis [Siller-Lopez et al. 2004; Iimuro et al. 2003]. Halofuginone is a coccidiostat that increases fibrolytic MMP expression [Ohayon et al. 2008]. Concurrently, inhibition of TIMP leads not only to enhanced activity of MMPs, but also to reduced HSC survival, which promotes apoptosis and clearance of the fibrogenic cells. Polaorezin downregulates TIMP-1 and TIMP-2 expression, and reportedly attenuates fibrosis [Sugino et al. 2008].
Stimulate stellate cell apoptosis
Myofibroblast clearance by apoptosis is one of the key features of the liver’s endogenous response to remove scar. The relative apoptotic activity of HSCs reflects a balance between apoptotic stimulation and survival signals, which can be manipulated therapeutically.
Interactions between HSCs and the surrounding matrix influence the propensity towards apoptosis. Degradation of the fibrotic matrix and especially collagen I and TIMP-1 reduces survival signals for activated HSCs. Increased survival of HSCs may result from increased expression of anti-apoptotic proteins such as Bcl-2 [Novo et al. 2006], and by transcription factors, especially NF-κB. NF-κB is a pro-inflammatory transcription factor, which is constitutively active in myofibroblasts, and reduces sensitivity toward apoptotic signaling, thereby promoting survival. Inhibition of NF-κB by gliotoxin, a fungal product, accelerates recovery from fibrosis in an animal model [Pahl et al. 1996], which has encouraged the use of sulfasalazine as a potential drug because of a similar mechanism of action [Oakley et al. 2005]. ACE inhibitors also reduce myofibroblast survival by upstream inhibition of NF-κB signaling [Oakley et al. 2009]. Increased myofibroblast apoptosis also results from the use of either a CB1 antagonist (rimonabant) or a 5HT antagonist [Ruddell et al. 2006]. The challenge is to specifically target myofibroblast survival without damaging hepatocytes or stimulating the immune system.
Another key cell type involved this process are NK cells, which are a vital component of the innate immune system that are potentially important antifibrogenic effectors in the injured liver. Thus, activation of NK cells could be a novel, therapeutic target to treat liver fibrosis [Gao et al. 2009]. When HSCs adopt the activated phenotype they become susceptible to NK cell-induced apoptosis, in part through downregulation of the inhibitory NK cell ligand and upregulation of TRAIL receptors [Radaeva et al. 2006]. In addition, production of IFNγ, a hallmark of NK cell activation, is another important mechanism contributing to the antifibrotic effects of NK cells. IFNγ not only inhibits HSC activation directly, but also amplifies NK cell cytotoxicity against HSCs via upregulation of NKG2D and TRAIL expression on NK cells [Jeong et al. 2006]. Accordingly, a pilot study using IFNγ reduced fibrosis in selected HCV infected patients [Muir et al. 2006]. In a randomized, open-labeled, multicenter trial of IFNγ in patients with HBV infection, hepatic fibrosis scores were significantly reduced in 63% of IFNγ treated patients compared with 24.1% in the control group. Semiquantitative fibrosis scoring was decreased from 13.8 ± 5.8 to 10.1 ± 5.1 percent in the IFNγ group (p = 0.0001), whereas they were unchanged in control subjects [Weng et al. 2005].
Bone marrow transplantation
Use of BM progenitor cells (BMCs) to promote regeneration and enhance matrix degradation is an intriguing and somewhat controversial new approach to antifibrotic therapy. Several signaling pathways linking progenitor cell activation and myofibroblasts have been identified, and there is increasing evidence that crosstalk (both physical and via soluble factors) between those cell types is essential for both fibrosis and parenchymal regeneration. Therefore, from the therapeutic perspective, cirrhosis with chronic liver failure is an appealing target disease. Furthermore, BMCs may degrade liver matrix (fibrolysis) by increasing the expression of MMPs [Sakaida et al. 2004].
In animal models, BM transplantation improves liver function and ameliorates hepatic fibrosis [Roderfeld et al. 2010; Sakaida et al. 2004]. Moreover, preliminary data about safety and tolerability are encouraging. In a small group of patients receiving autologous transplantation of CD133 + mesenchymal stem cells to repopulate the liver after extensive hepatectomy, there was a 2.5-fold increased mean proliferation rate of their left lateral segments [am Esch et al. 2005].
Ongoing clinical trials are investigating stem cell and mesenchymal stem cell transplantation using adult or fetal liver, or umbilical cord as sources of progenitor cells in a range of diseases via different delivery techniques including peripheral, intraportal, intra-arterial administration (Table 1 and ClinicalTrials.gov). As these efforts unfold, it will be essential to define exactly which cells confer antifibrotic activity, and to develop safe, reproducible methods to isolate, characterize and administer them.
Other approaches
A number of studies implicate the coagulation system in the modulation of fibrosis. There have been a few reports of accelerating fibrosis in patients with procoagulant factor V Leiden mutation and HCV infection [Wright et al. 2003]. In a mouse model of factor V Leiden, a rapid fibrosis rate was also observed, which was reduced significantly using anticoagulant therapy. Furthermore, thrombin activates HSCs through the PAR1 receptor [Wright et al. 2003]. These studies have generated the hypothesis that liver injury promotes fibrosis by activating the coagulation system, thereby generating thrombin which activates HSCs. Therefore, anticoagulation may attenuate fibrosis and delay or prevent the development of end-stage liver disease (see ClinicalTrials.gov).
The anti-inflammatory properties of insulin therapy, along with its ability to reduce insulin resistance over time have led to the use of insulin therapy in patients with chronic HCV liver cirrhosis (see ClinicalTrials.gov)
Summary
In 2003 [Friedman, 2003], we described the vision of the ideal assessment and therapy for a 50-year-old male with chronic HCV in 2012:
“The physician will use his palm-sized digital assistant to calculate a fibrosis risk score based on a combined index… Next, a non-invasive assessment of fibrosis will be obtained using a novel imaging modality that quantifies fibrogenesis, combined with a multiplex serum assay. If all indices indicate high risk of fibrosis, a customized multi-drug antifibrotic regimen…will be initiated. The patient will live to his natural life expectancy free of end-stage liver disease or its complications.”
A decade later, maybe it was too optimistic to achieve this goal by 2012, but it is clear that we are getting closer. Several noninvasive techniques are already in clinical use, reducing the need for biopsy. In particular, serum tests and transient elastography predict the risk of decompensation better than liver biopsy, further justifying their eventually replacing biopsy in antifibrotic trials and clinical management. The assessment of the fibrosis risk score is not yet by ‘palm-sized digital assistant’, but many of those variables could be incorporated into a decision-making framework that determines how urgently therapy is needed. Slowly but steadily, conditions are aligning that will eventually yield successful antifibrotic therapies. It is difficult to predict which therapies will succeed first, but existing drugs with established safety records are likely to be developed fastest.
Footnotes
Funding
Dr. Friedman is supported by funding from NIH (DK56621) and a research grant from Onyx Pharmaceuticals.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.