
Abstract
Background
Epigenetic modifications such as DNA methylation in the CpG islands of genes occur at a high rate. In this study, we measured the methylation level of the promoter region of the FOXF1 gene as a new blood biomarker for the detection of colorectal cancer in the early stages.
Methods
The methylation level of the promoter region of the FOXF1 gene was measured in the plasma samples of 50 colorectal cancer patients and 50 normal individuals. DNA was extracted after exposure to sodium bisulfite by the MethyLight polymerase chain reaction (PCR) method. The percentage of promoter region was measured in all samples, and statistical analysis was done using SPSS v24 software.
Results
The average promoter region between the plasma samples of colorectal cancer patients and healthy individuals had a significant difference (P < 0.001). The average promoter region of the FOXF1 gene in tumor plasma samples was 7.1 and in the control samples was 0.48. The sensitivity and specificity of the sample plasma levels were 78% and 89.5%, respectively.
Conclusion
The promoter region value of the FOXF1 gene in plasma samples using the MethyLight PCR method had high sensitivity and specificity as a non-invasive method for colorectal cancer diagnosis. This research is the first report that has been presented regarding the investigation of FOXF1 gene methylation in plasma samples in colorectal cancer. Therefore, it is necessary to conduct more studies with larger size samples to evaluate the efficiency of the gene under investigation.
Introduction
According to the GLOBOCAN 2020 cancer incidence and mortality estimates, colorectal cancer (CRC) is the third most diagnosed cancer. However, after lung cancer, which remains the leading cause of cancer death, CRC was the second leading cause of cancer death. 1 However, CRC progresses slowly; by screening and diagnosis in the early stages, the reduction of mortality due to this type of cancer is fulfilled. Among the main reasons that limit the screening of this disease are the lack of access to diagnostic laboratories, limitations of test performance, and imperfect screening conditions. 2 For a long time, researchers have been looking for non-invasive diagnostic ways to replace colonoscopy, which is still the gold standard for CRC screening and diagnosis. They wanted to increase patient participation and bypass the obstacles of bowel preparation for diagnostic colonoscopy tests. 3 Various CRC screening tests based on the blood and other body fluids were introduced2,4 besides stool bacterial and metagenomics markers, fecal proteins, genetic, and epigenetic markers in stools. 3
Cancer is caused by the cooperation of genetic factor (age, mutagenic chemicals, UV light) and epigenetic factor (molecular factors and processes around DNA that regulate gene expression independent of DNA sequences) changes. Most of the changes are transient, but their accumulation causes cancer. One of the epigenetic changes created is aberrant DNA methylation. With a combination of aberrant DNA methylation and point mutations, the risk of developing cancer is predictable. 5 DNA methylation is an enzymatic phenomenon that occurs in CpG islands, a region rich in cytosine and guanine. CpG islands are concentrated in the promoter regions of 60–70% of genes and are not methylated in the normal state; unusual methylation of this promoter region causes silence of gene expression.6,7 Mitchell et al. 8 investigated DNA methylation in 23 genes of blood and tissue of CRC patients compared with healthy subjects. They reported that 11 genes (BCAT1, COL4A2, DLX5, FGF5, FOXF1, FOXI2, GRASP, IKZF1, IRF4, SDC2, and SOX21) had low methylation in peripheral blood DNA and were suitable for further evaluation as blood-based CRC diagnostic markers. In screening for early detection of CRC, the US Food and Drug Administration (FDA) has approved three DNA methylation markers, including SEPTIN9, NDRG4, and BMP3. However, only 0.8% of methylation markers have been converted into a commercial product, indicating the complexity of translating laboratory discoveries into clinical application, which can be due to study flaws, unfavorable markers, or methodological flaws. 9
Previous studies in various cancers suggest that the role of FOXF1 in tumorigenesis is complex. FOXF1 can induce metastatic breast cancer cells through the induction of epithelial-mesenchymal transition (EMT; the biological process in which polarized epithelial cells transform into cells with a mesenchymal phenotype), up-regulation of lysine oxidase, down-regulation of SMAD2/3, and activation of the MAPK pathway. 10 On the other hand, hypermethylation of the FOXF1 promoter in breast cancer cell lines makes it a tumor suppressor. 11 In lung cancer fibroblasts, promoting the expression of hepatocyte growth factor can play a role in the aggressive metastasis of lung cancer fibroblast cells. 12 In hepatocellular cells, FOXF1 acts as an inhibitor of cell invasion and tumorigenesis. 13 Also, it was revealed that both high expression and nuclear/cytoplasm localization of FOXF1 were positively associated with aggressive phenotype and poorer outcomes of CRC, FOXF1 might contribute to the progression and metastasis of CRC. 14 In the study of Wang et al., 14 FOXF1 increased expression was associated with angiogenesis in CRC. They concluded that FOXF1 acts as a promotor for the transcription of vascular endothelial growth factor A1 and changes tumor progress. They reported that the higher level of FOXF1 was positively associated with an enrichment of EMT gene signatures, and exogenous overexpression of FOXF1 induces EMT by transcriptionally activating SNAI1. Lo et al. 15 have reported that FOXF1 expression in CRC cell lines with inactive P53 and knockdown of FOXF1 provokes genomic instability in CRC cells in FOXF1-expressing CRC cells with a defect in the p53-p21 (WAF1) checkpoint, indicating that FOXF1 is an essential element in the colorectal tumor. In another study, they showed that the expression of FOXF1 cytoplasmic protein in epithelial cells of the tumor has a positive correlation with the degree of histology, the depth of the invasion, the stage, and the lymphatic metastases of the colorectal adenocarcinoma. 16
Considering the role of FOXF1 in CRC, it is possible to use it as a diagnostic biomarker by examining epigenetic events in this gene. So far, there has been no investigation related to the quantification of FOXF1 gene promoter methylated DNA in blood samples as a diagnostic marker of CRC. In this study, the presence of the FOXF1 gene promoter methylated DNA in the plasma of CRC patients (by the MethyLight polymerase chain reaction (PCR) method) is compared with healthy samples. This study was conducted to determine whether the quantity of circulating methylated DNA of the FOXF1 gene is different in healthy individuals and CRC patients. In addition, we compared the level of a typical tumor marker carcinoembryonic antigen (CEA) 17 with the FOXF1 methylated DNA in plasma samples.
Material and methods
Study design and samples
This study is part of a project that has been approved by the Ethics Committee of the Faculty of Medicine of Kermanshah University of Medical Sciences (project reference 97223), and written consent has been obtained from all participants in this study.
In a case-control study, 50 patients with CRC (patient group) and 50 healthy subjects (controls) were included, and after blood sampling, their plasma was separated. The control group was selected by matching the age and gender of the subjects. The studied population included people who had been referred to Mahdia Clinic and Imam Reza Hospital, Kermanshah, Iran. The inclusion criteria of patients included any age group, both sexes, and a range of cancerous tumor characteristics (histological differentiation degree, tumor location, tumor size, disease stage) that confirmed the presence of a cancerous lesion in the pathological report after colonoscopy and sigmoidoscopy. Patients with a history of chemotherapy, radiotherapy, and tumor surgery—and those who did not agree to enter the study—were excluded. The control group was selected from the employees of the same medical centers. The inclusion criteria of the control group were: (a) no report of abnormal pathology after colonoscopy; (b) no history of malignancy and underlying systemic disease; (c) a negative fetal occult blood test (d) normal blood CEA; and (e) no inflammatory disease.
Sample collection, processing, and storage
Blood sampling (10cc) was collected from 50 healthy subjects and 50 CRC patients. The sampling was done under standard conditions; blood was drowned with a Venoject needle and transferred in tubes containing an EDTA anticoagulant. To obtain the plasma, the samples were centrifuged for 15 min (2500 rpm, 4°C), then allocated in sterile microtubes DNase-free and RNase-free and stored at −80°C for further tests.
DNA extraction from plasma samples
Whole genome DNA was extracted from 200 µL plasma. DNA extraction was performed according to the instructions of the DNA extraction kit (Qiagen, Cat. No. 51304, Germany). DNA was eluted in buffer AE (Qiagen GmbH, Germany) and stored at −20°C.
Bisulfite modification
The essential step for the correct detection of the DNA methylation pattern is exposure to sodium bisulfite. Non-methylated cytosine undergoes deamination in the vicinity of sodium bisulfite and turns into uracil, but methylated cytosine remains unchanged. 18 This process is accomplished in two steps. First, to convert non-methylated cytosine to uracil, DNA incubates in a high concentration of bisulfite salt, low pH, and high temperature. Second, the DNA purifies to remove bisulfite and other chemicals.
In the DNA treatment with sodium bisulfite step, according to the instructions of the kit manufacturer (Qiagen, Cat. No 59104, Germany), 40 µL of extracted DNA, 85 µL of bisulfite mix, and 25 µL of DNA protect buffer were mixed. Bisulfite treatment was performed according to the following protocol:
Initial denaturation (95°C, 5 min); incubation (60°C, 15 min); second denaturation (95°C, 5 min); incubation (60°C, 15 min); and keeping at 20°C for 10 min.
After the bisulfite modification stage, DNA purification was performed, which briefly included:
560 µL of carrier RNA dissolved in BL buffer (10 µg/mL), mixed, and spun. Carrier RNA increases the binding of DNA to the MinElute DNA spin column membrane. Washing with BW washing buffer (Qiagen, GmbH, Germany) Sulphonation of membrane-bound DNA Washing with desulphonation buffer (Qiagen, GmbH) Elution of the pure converted DNA from the spin column
The quantitative of the DNA is evaluated by measuring the absorption of UV light (260 nm) by the aromatic arms of DNA. Also, the protein and RNA contamination are detected by measuring their absorption at the wavelength of 260/280 nm and 230/280 nm respectively
MethyLight PCR method
The MethyLight PCR is a TaqMan-based method that is utilized to detect the methylated sequence of the promoter region of the FOXF1 gene in bisulfite-treated DNA samples with a high sensitivity.
To perform MethyLight PCR, the EpiTect MethyLight PCR + ROX vial kit (Qiagen, Cat. No 59496, Germany), a pair of primers, and a TaqMan probe designed by Beacon Designer™ (version 8.13; www.premierbiosoft.com/molecular_beacons; Premier Biosoft International, Palo Alto, CA, USA) are applied.
The ALU-C4 reference gene was employed to normalize the amount of input DNA in the PCR reaction; to amplify the reference gene; a pair primer and a probe designed for the ALU-C4 sequence independently of the methylation status (lacking CpG dinucleotide) were utilized.
Designing the probe and primers
The sequence of the promoter region of the FOXF1 gene was extracted from reliable databases such as USCS Genome Browser (http://genome.ucsc.edu/). The CpG island-specific primers and probes for their fully methylated state were designed using Beacon Designer™ software. Appropriate primers and probes were determined according to the general conditions of primer and probe design (Table 1). The possibility of the formation of a hetero dimer, hairpin, and self-dimer was also checked using the Oligo Analyzer tool. To ensure the specificity of the designed primers, the primers were blasted in NCBI. Primers and probes designed by Metabion (Steinkirchen, Germany) were synthesized. The length of the FOXF1 gene promoter amplicon in the database (Gene Bank: NM 001451) was 118 bp. This region contains 18 base pairs of CpG, which covers the forward primer of 3 base pairs of CpG, the reverse primer of 1 base pair of CpG, and the probe of 3 base pairs of CpG.
Primer and probe sequences used for MethyLight polymerase chain reaction.
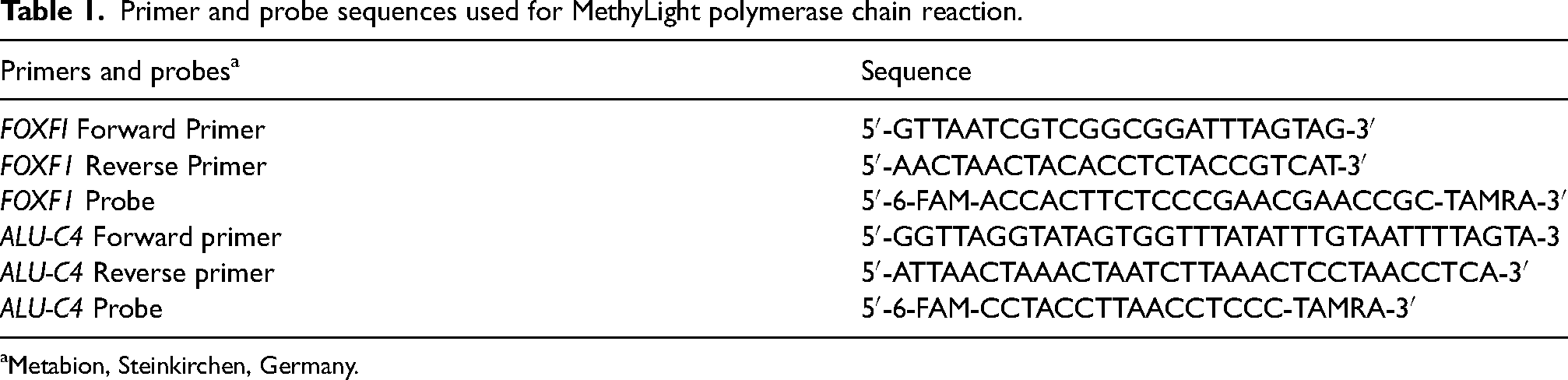
Metabion, Steinkirchen, Germany.
MethyLight quantitative PCR
Quantitative PCR (qPCR) reactions were performed by PCR 7500 Real-Time (Applied Biosystems; Thermo Fisher Scientific, Inc.), using PCR + EpiTect Methylight PCR + ROX Vial Kit containing Master Mix and ROX Dye Solution (Qiagen, Germany) according to the manufacturer's protocol. These reactions in a volume of 20 µL include 2X EpiTect MethyLight Master mix (without ROX) (10 µL), 2 µL of 10× primer-probe mix (0.4 µM forward primer, 0.4 µM reverse primer, 0.2 µM probe), 0.4 µL of 50× ROX Dye Solution, 1 µL bisulfite-treated DNA template (concentration <100 ng/optimum reaction), and RNase-free water (6.6 µL).
The cycling conditions were as follows: initial PCR activation step at 95°C for 5 min, 50 cycles at 95°C for 15 s (denaturation step), and 59°C for 1 min (annealing/extension step with fluorescence data collection).
Reactions were performed in a 96-well plate, and each plate contained standard curves for FOXF1 and ALU-C4 sequences, human methyl bisulfite-modified DNA as a positive control, DNA modified with unmethylated human bisulfite as negative control (Qiagen, Cat. No 59655, Germany), unmodified human genomic DNA from control samples that were not amplified, and no template control (containing all reaction components except template DNA).
Real-Time PCR software (ABI 7500 SDS v1.3.1) was used to analyze and collect the results in the real-time PCR stage. This software calculated the cycle threshold (Ct) value for each sample when the fluorescence level exceeded the threshold. The value of Ct is inversely proportional to the value of input DNA concentration, and the magnitude of fluorescence produced is directly proportional to the PCR product. We acquired a proper MethyLight PCR outcome where the PCR efficiency (92–100%), correct slope value (3.1–3.5), and R2 = 0.956–0.999. In this research, a 100% methylated bisulfite commercial DNA template was utilized as a positive control to calculate the percentage of methylated ratio (PMR) in unknown samples.
PMR is the methylation percentage of the ALU-C4 gene in each sample compared to the positive control commercial sample with 100% methylated DNA. The concentration of methylated DNA (related to the FOXF1 gene) and the concentration of reference gene DNA were calculated by the standard curves. In the standard curve, the logarithmic concentration of input DNA was plotted against Ct. normalized values were calculated for each sample by dividing the value obtained of the FOXF1 gene on the ALU-C4 gene. To calculate the PMR, the normalized value of each sample was divided by the normalized value of commercial methylated human bisulfite-converted DNA, and the result was reported as a percentage. The formula used in the calculations is as follows: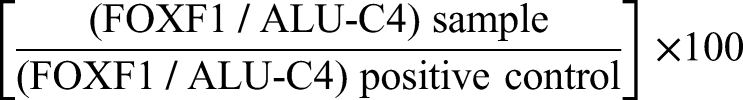
CEA plasma level assay
CEA level was measured by Liaison® chemiluminescence analyzer (Liaison CEA kit, Italy) in all control and case samples according to the protocol of the manufacturer of the kit.
FOBT
The FOBT was performed in stool samples of the control and case groups using the alcohol pyrimidine method. Its mechanism is changing the color of alcohol pyrimidine by blood peroxidase in feces. In this way, by adding H2O2 to the stool sample, if there is blood, the peroxidase enzyme causes the decomposition of hydrogen peroxide and the release of oxygen, which is oxidized by pyrimidine alcohol and changes the color (usually blue to purple). The intensity of the color change has a direct relationship with the activity of the peroxidase enzyme or the content of blood in the feces.
Statistical analysis
The data were statistically analyzed using SPSS v24.0 software. At first, based on the results of the Kolmogorov–Smirnov statistical test, the normal distribution of the sample was rejected. Then the Mann–Whitney and Chi-square tests were used to analyze the results, and in all the tests, the P-value was <0.05. The Mann–Whitney test was used to compare the concentration of methylated DNA in the promoter region of the FOXF1 gene in stool and plasma samples. To determine the sensitivity and specificity of the methylation level of the FOXF1 in the diagnosis of CRC, the receiver operating characteristic (ROC) 19 was plotted.
Results
Demographic and clinicopathologic results
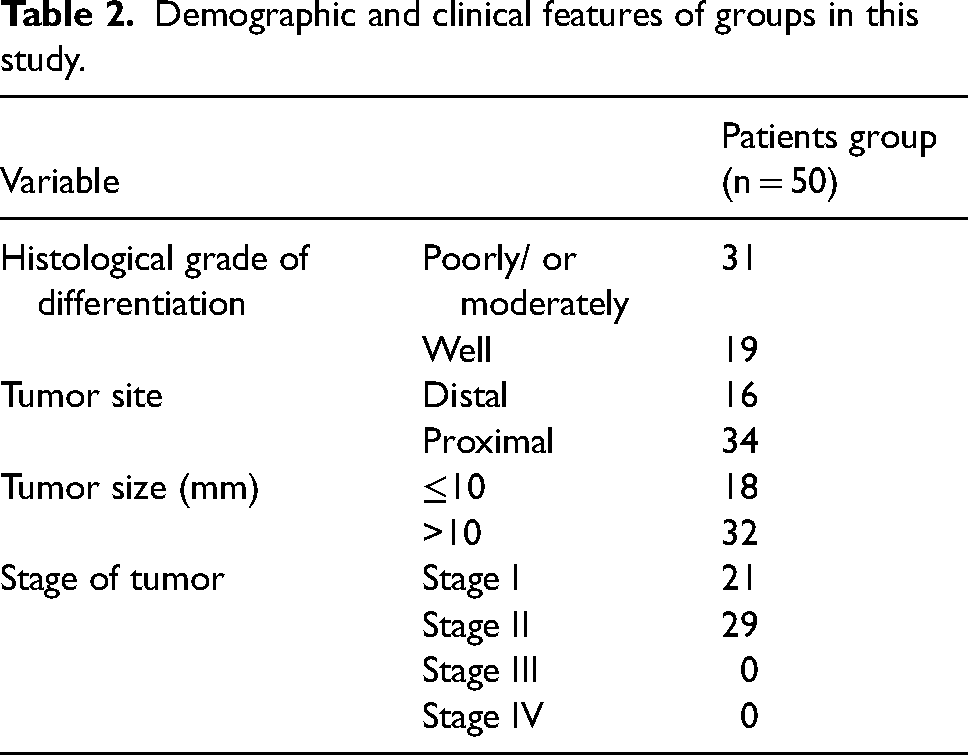
In general, 50 blood samples were taken from patients with CRC and 50 samples were taken from the control group. The average age of the patients was 56 ± 12 years old and there were 30 women and 20 men. The control group was matched with the patient group in terms of age (54 + 12) and sex (26 women and 24 men). The clinical characteristics of the study participants are given in Table 2.
Demographic and clinical features of groups in this study.
Concentration of methylated DNA (PMR) in the FOXF1 gene promoter region in plasma samples
The value of Ct of case and control groups was analyzed and calculated by real-time PCR device software. Then, the concentration of methylated DNA in the promoter region of the FOXF1 gene in the samples was calculated according to the PMR, and the relationship between the methylation status (positive or negative) and the demographic and pathological characteristics of the patients was investigated. The value of PMR in the plasma samples of patients and control was 7.1 and 0.48, respectively. The highest value of PMR was related to the plasma of the control group, which was considered the threshold of methylation status. The PMR value of the patient group was significantly higher than the PMR of the control group, and there was a significant difference between the two groups (P < 0.001). There was no statistically significant relationship between the PMR value in the patient plasma samples and the demographic and pathological characteristics of the patients (Table 3).
Value of PMR FOXF1 gene in plasma of patients with CRC and its relationship with clinicopathological characteristic.
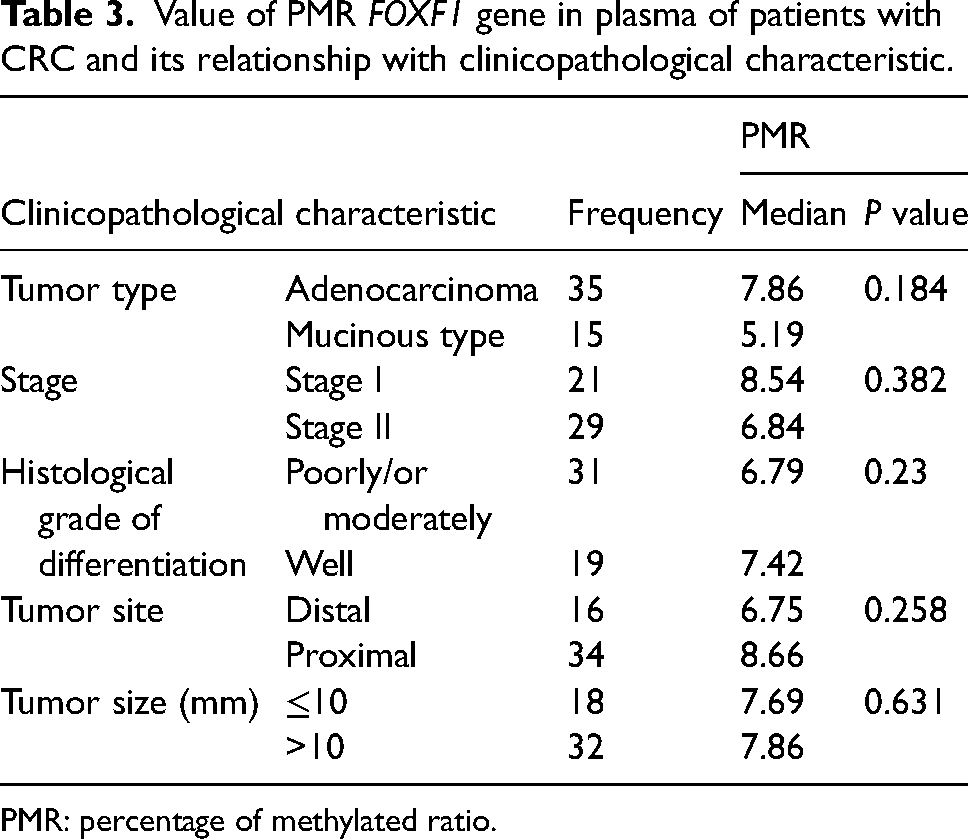
PMR: percentage of methylated ratio.
Also, there was no statistically significant difference between the FOXF1 gene methylation status in patient plasma samples and the demographic and pathological characteristics of the patients (Table 4).
Frequency DNA hypermethylation of FOXF1 in plasma of CRC patients and its relationship with clinicopathological characteristic.
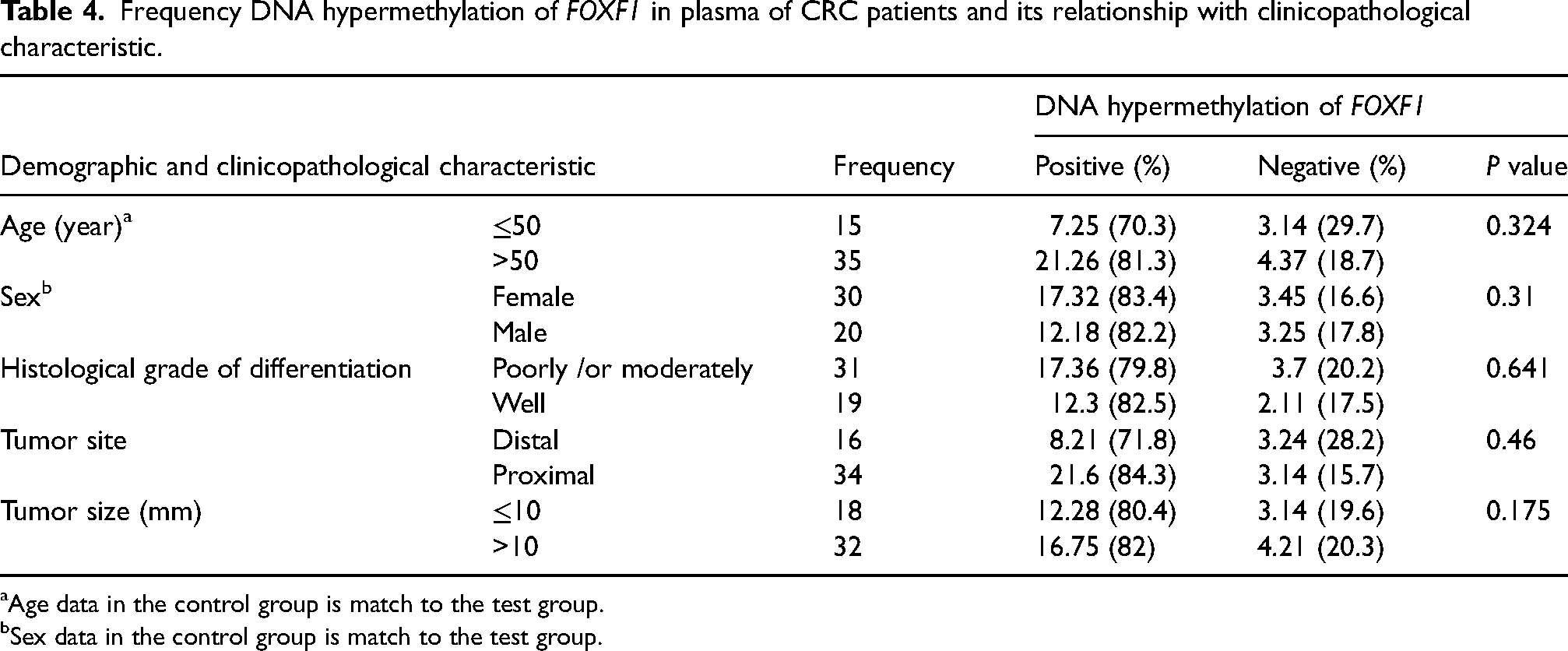
Age data in the control group is match to the test group.
Sex data in the control group is match to the test group.
Sensitivity and specificity of FOXF1 gene PMR index as blood biomarker in CRC diagnosis
Using SPSS software and analyzing the results by ROC curve, the sensitivity and specificity of the FOXF1 gene PMR index in plasma were determined. The highest value of PMR was related to the plasma of the control group (2.92), which was considered the threshold of methylation status. Based on this threshold, 43/50 of patients showed positive methylation status. According to the ROC curve, the area under the curve shows the efficiency of the FOXF1 gene PMR assay in plasma samples as a diagnostic method, which has 78% sensitivity and 89.5% specificity (Figure 1(a)).
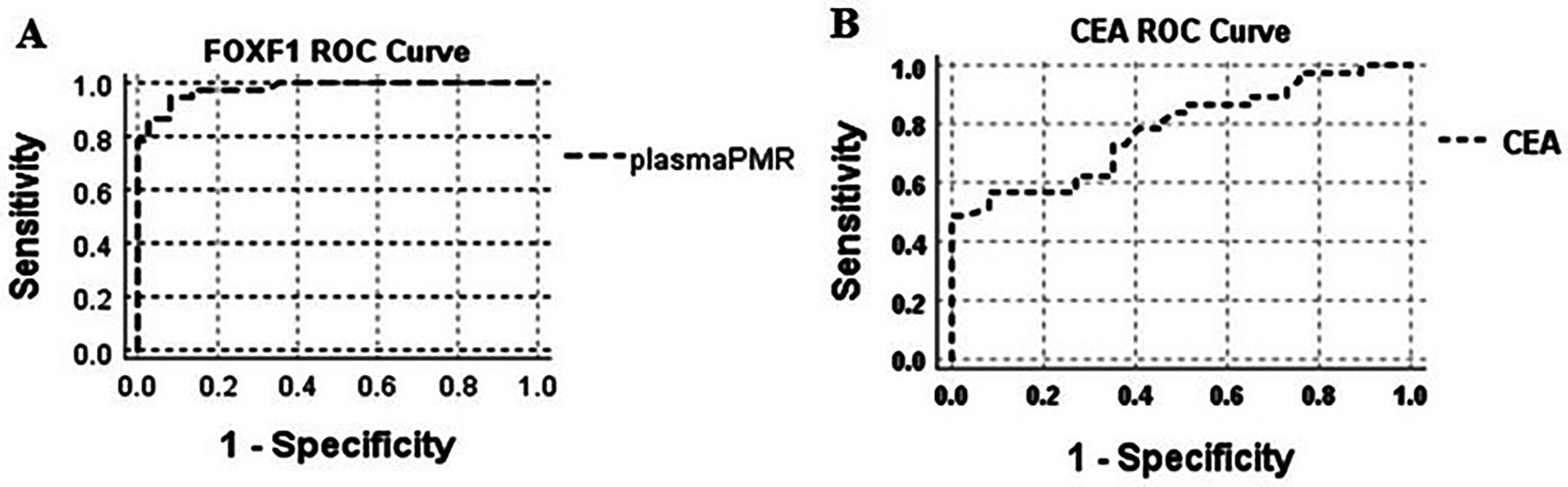
Receiver operating characteristic curve for PMR in plasma samples of colorectal cancer patients. The area under the curve shows the efficiency of FOXF1 gene PMR assay in plasma samples as a diagnostic biomarker for colorectal cancer (a). Receiver operating characteristic curve for CEA in plasma samples of colorectal cancer patients (b). CEA: carcinoembryonic antigen; PMR: percentage of methylated ratio.
Level of CEA plasma and FOBT results
The statistical analysis of CEA levels in the plasma samples of CRC patients showed a statistically significant difference in these results compared to the plasma samples of healthy people as a control group (P < 0.05). The value of CEA, according to the median index in the plasma samples of patients, was 3.74 and in the control was 0.97. Regarding the CEA level in the plasma samples of patients, a sensitivity of 41% and specificity of 98% was evident (Figure 1(b)).
After conducting an occult blood test in each of the stool samples 27/50 patients were positive for FOBT.
Discussion
This case-control study was conducted to achieve an early diagnosis of CRC by examining the promotor methylation rate of the FOXF1 gene in plasma samples of CRC patients. In this study, we used the MethyLight PCR method as a high-performance quantitative method to measure the methylation level in bisulfite-treated DNA, and the PMR value for the FOXF1 gene was measured in the plasma of CRC patients. The results of the ROC curve analysis showed that according to the sensitivity of 78% and the specificity of 89.5%, the PMR value of the FOXF1 gene is likely a blood biomarker in the diagnosis of CRC. However, there was no significant difference in the value of this index among variables such as tumor type, stage of the tumor, histological grade of differentiation, tumor site, and tumor size in CRC.
Achieving non-invasive and early detection methods in CRC is one of the topics on which various research has been conducted. In this regard, stool, serum, and blood plasma samples were employed to check biomarkers and diagnostic tools. The multi-target stool DNA test (Cologuard, a combination of NDRG4 and BMP3 DNA methylation, and hemoglobin), plasma SEPT, and DNA methylation test (Epi pro colon) are among the non-invasive methods that have been approved by the FDA.20,21 However, these methods are not satisfactory in terms of economic efficiency and diagnostic accuracy. Therefore, along with these approved methods, fecal/blood-based microRNA and CRC-related gut microbiome screening markers have also been investigated to provide the best molecular biomarkers with high screening sensitivity and specificity. 22
Previous studies have shown that although genetic changes play a prominent role in a subset of CRC, the contribution of epigenetic aberrations in this malignancy is also significant. Since epigenetic effects occur in the early stages of disease pathogenesis, they include almost all critical pathways related to cancer and may utilize as disease-related biomarkers in diagnosis, treatment prediction, and prognosis. In this regard, DNA methylation, histone modifications, and the role of non-coding RNAs are significant as epigenetic regulators. 23 DNA methylation modifications (hypomethylation and hypermethylation) that occur in the standard unmethylated regions and CpG islands of the gene promoter induce different consequences for the cell. It has been suggested that CpG promoter hypermethylation suppresses the transcription of tumor suppressor genes in cancer cells. 24 In the case of CRC, aberrant hypermethylation has been detected in CDKN2A, hMLH1, and APC19 gene promoters, as well.19,25
These findings led the researchers to investigate and introduce diagnostic biomarkers according to the methylation status of different genes. To identify plasma molecular markers in CRC diagnosis, Lee et al. 26 examined 10 genes in the tissue and plasma of 243 patients and 276 healthy subjects. The methylation score in a model including APC, MGMT, RSSF2A, and Wif-1 genes had a sensitivity of 86.5% and a specificity of 92.1%. In this model, the methylation score had a positive predictive value of 90.6% and a negative predictive value of 88.8%. Pederson et al. 27 investigated the specific genes BCAT1and IKAZF1, which have high methylation in CRC tissue compared to healthy tissue. They isolated plasma samples from 2105 volunteers and identified CRC in 85 out of 129 cases using the studied genes (66% sensitivity; 95% CI 56, 74). For stages I–IV of CRC, the positive rate was 38%, 69%, 73%, and 94%, respectively; that is, an increasing trend was observed between positivity and invasiveness. In addition, there was a decrease in the methylation signal after tumor surgery. However, tumor site, sex, age, smoking, and family history do not affect the positive rate of the test. The sensitivity of these markers in advanced adenoma was 6%. In a study, SDC2 gene methylation was considered a potential biomarker for early diagnosis of CRC. In clinical validation, a high level of aberrant SDC2 methylation was measured in the tissues of 139 CRC patients in most primary tumors (97.8%). Clinical validation of SDC2 methylation in DNA of CRC patients (n = 131) in stages I–IV with the qPCR methylation method showed >87% sensitivity and 95.2% specificity. Interestingly, the sensitivity was 92.3% in stage I, which means that SDC2 methylation can potentially be a blood-based DNA test for early detection of CRC. 28 Rezvani et al. 29 investigated the value of methylated DNA of SPG20 gene promoter in plasma and tissue samples of CRC patients by the MethyLight PCR method. They showed that the percentage value of PMR in the plasma and tissue samples of CRC patients was higher than the healthy subjects. They reported a sensitivity of 81.1%, a specificity of 96.9% for plasma samples, a sensitivity of 93.8%, and a specificity of 99.6% for tissue samples according to the methylation level of the SPG20 gene promoter.
Also, the potential of LINE-1 gene hypomethylation as a biomarker for CRC diagnosis was investigated. The level of hypomethylation of the LINE-1 gene in circulating cell-free DNA (cfDNA) in 114 CRC patients was investigated by real-time PCR assay for quantitative analysis of methylated alleles. It was suggested that CRC patients with tumor size >6 cm, advanced, and metastatic stages have statistically higher cfDNA LHI than other CRC patients. In addition, stage I compared to stage II, and stage III compared to stage IV had higher cfDNA LHI. Therefore, the cfDNA LHI is a worthwhile biomarker to predict the progression of CRC disease. 30
In this study, we measured the sensitivity and specificity of the CEA in patient samples and compared it with the PMR value of the FOXF1 gene; it had lower sensitivity and higher specificity.
According to the results obtained in this study, it is likely that the measurements PMR value of the FOXF1 gene in the plasma samples of CRC patients who were in stages I and/or II of the disease has the specificity of an early diagnostic biomarker. However, for the final confirmation, more studies are needed with higher sample sizes and different stages of the disease. In addition, the economic benefit of using this biomarker should be investigated in comparison with those that have been approved by the FDA.
Conclusion
PMR value of the FOXF1 gene in plasma samples using the MethyLight PCR method had high sensitivity and specificity as a non-invasive method for CRC diagnosis. This research is the first report that has been presented regarding the investigation of FOXF1 gene methylation in plasma samples in CRC. Therefore, it is necessary to conduct more studies with larger size samples to evaluate the efficiency of the gene under investigation.
Footnotes
Author contributions
ZD: literature search, laboratory work, data acquisition, data analysis, manuscript preparation, manuscript editing, manuscript review; NR: concepts, design, definition of intellectual content, clinical studies, manuscript preparation, manuscript editing, manuscript review, data acquisition, guarantor; SA: concepts, data acquisition, statistical analysis, manuscript preparation, manuscript review.
Consent to participate
Written informed consent was obtained before the interviews.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
This study is part of a project that has been approved by the Ethics Committee of the Faculty of Medicine of Kermanshah University of Medical Sciences (project reference 97223).
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.