
Abstract
Background:
Despite effective treatments, metastatic colorectal cancer (mCRC) prognosis is still poor, mostly in RAS-mutated tumors, thus suggesting the need for novel combinatorial therapies. Epigenetic alterations play an important role in initiation and progression of cancers, including CRC. Histone-deacetylase inhibitors (HDACi) have shown activity in combination with chemotherapy in the treatment of solid tumors. Owing to its HDACi activity and its safe use for epileptic disorders, valproic acid (VPA) is a good candidate for anticancer therapy that we have largely explored preclinically translating our findings in currently ongoing clinical studies. We have shown in CRC models that HDACi, including VPA, induces synergistic antitumor effects in combination with fluoropyrimidines. Furthermore, unpublished results from our group demonstrated that VPA induces differentiation and sensitization of CRC stem cells to oxaliplatin. Moreover, preclinical and clinical data suggest that HDACi may prevent/reverse anti-angiogenic resistance.
Methods/Design:
A randomized, open-label, two-arm, multicenter phase-II study will be performed to explore whether the addition of VPA to first line bevacizumab/oxaliplatin/fluoropyrimidine regimens (mFOLFOX-6/mOXXEL) might improve progression-free survival (PFS) in RAS-mutated mCRC patients. A sample size of 200 patients was calculated under the hypothesis that the addition of VPA to chemotherapy/bevacizumab can improve PFS from 9 to 12 months, with one-sided alpha of 0.20 and a power of 0.80. Secondary endpoints are overall survival, objective response rate, metastases resection rate, toxicity, and quality of life. Moreover, the study will explore several prognostic and predictive biomarkers on blood samples, primary tumors, and on resected metastases.
Discussion:
The “Revolution” study aims to improve the treatment efficacy of RAS-mutated mCRC through an attractive strategy evaluating the combination of VPA with standard cancer treatment. Correlative studies could identify novel biomarkers and could add new insight in the mechanism of interaction between VPA, fluoropyrimidine, oxaliplatin, and bevacizumab.
Trial Registration:
EudraCT: 2018-001414-15; ClinicalTrials.gov identifier: NCT04310176
Background
Despite effective treatments, metastatic colorectal cancer (mCRC) prognosis is still poor, mostly in RAS-mutated tumors, 1 where intrinsic cancer biology factors suggest the need for novel combinatorial therapies. This project will address this clinical need and could lead to the availability of a novel and affordable treatment. In RAS-mutated mCRC setting, the combination of chemotherapy and anti-VEGF bevacizumab is the only therapeutic option in first line. 2 We recently completed a phase-III study (OBELICS trial, grant RF-2009-1539464) 3 in 230 mCRC patients, investigating different schedules of bevacizumab plus oxaliplatin/fluoropyrimidine regimens (mFOLFOX-6/mOXXEL), and confirmed the worst outcome of RAS-mutated patients.
National health systems, particularly in Italy, are unlikely to support the explosion in costs of new oncology drugs for much longer. In light of this situation, the optimization of consolidated anticancer drugs as well as the mechanistic-based repurposing in cancer treatment of cheap and safe non-anticancer drugs already in clinical practice, represent attractive strategies to offer more effective and affordable treatments to cancer patients.
HDAC inhibitors as anticancer agents
It has become evident that target-based anticancer agents often exhibit limited clinical activities. Biological redundancies and alternative pathways can often bypass the inhibition of a single target, suggesting that in some cases broad-specificity compounds, or multi-target drug therapies, may be more effective than individual high-affinity, high-specificity therapies. Epigenetic alterations, such as hypoacetylation of histones, play an important role in initiation and progression of several cancers, including CRC. Since epigenetic alterations are dynamic and generally reversible, epigenetic manipulation has emerged as an attractive novel anticancer treatment.
Histone deacetylases (HDACs) are chromatin modifiers enzymes regulating the acetylation of a variety of histone and non-histone proteins, controlling the transcription and regulation of genes involved in cell cycle control, proliferation, survival, DNA repair and differentiation, and their expression is frequently altered in hematologic and solid tumors. 4 Class I HDACs (HDACs 1–3 and 8) are predominantly expressed in the nucleus, and are the major mediators of histone deacetylation, whereas the class IIa HDACs (HDACs 4, 5, 7 and 9) and class IIb HDACs (HDACs 6 and 10) either shuttle between the nucleus and the cytoplasm, or are predominantly expressed in the cytoplasm and deacetylate non-histone proteins. 4
HDAC inhibitors (HDACi) are an emerging group of agents that, by targeting histone deacetylase, influence chromatin structure, thus regulating gene expression. Moreover, HDACi can also modulate cellular functions independent of gene expression by acting on non-histone proteins deacetylation, in this way being involved in the regulation of different altered pathway in cancer, such as apoptosis, cell cycle, and DNA repair. A large number of HDACi are currently in clinical development as anticancer agents, and three (vorinostat, romidepsin, and belinostat) have been approved by the US Food and Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma,5–7 whereas panobinostat is the first HDACi approved to treat recurrent multiple myeloma in combination treatment. 8 In solid tumors, including CRC, HDACi have failed to show considerable antitumor activity as single agent and are more active in combination with radiotherapy, chemotherapy, or other biological agents. 9
Valproic acid: preclinical and clinical studies
Valproic acid (VPA), an eight-carbon, branched-chained fatty acid, is a generic low-cost drug used for over 50 years as an anticonvulsant and mood stabilizer in the treatment of manic depression (bipolar affective disorder). Notably, VPA has HDAC inhibitory activity and anticancer properties. 10 Owing to its safe use as a chronic therapy for epileptic disorders, it could be considered a good candidate to be combined in novel anticancer regimens. Moreover, compared with other HDACi, VPA possesses better pharmacokinetics for clinical use with a substantially longer plasma half-life (7–9 h in humans). 11
Several phase-I and II clinical studies in hematologic and solid malignancies, including CRC, showed that VPA treatment, either as a monotherapy or combined with other agents, was reasonably well tolerated and resulted in some encouraging tumor responses.12–17 VPA was administered according to different schedules, most frequently orally at doses used for the treatment of epilepsy, either continuously during the whole duration of chemotherapy, or during a specified time of the chemotherapy cycle. The recommended values of serum concentrations for the treatment of epilepsy were in the 50–100 µg/ml range. As monotherapy, the maximum tolerated dose (MTD) of infusional VPA in solid tumors was 60 mg/kg/day for 5 consecutive days in a 21-day cycle, with neurotoxicity as the main dose-limiting toxicity (DLT) and no grade 3/4 hematological toxicity observed. 14
The ability of VPA to inhibit deacetylase activity in solid tumors has been demonstrated in monotherapy at oral doses between 20 and 60 mg/kg. 15 VPA oral doses of 30 mg/kg daily induced HDAC inhibition in the peripheral blood of patients in a neoadjuvant epigenetic therapy study in combination with the demethylating agent hydralazine plus doxorubicin and cyclophosphamide, for locally advanced breast cancer. In the latter study the mean plasma concentration was of 87.5 µg/ml, the therapy was safe, and tumor responses appeared higher as compared with historical controls. 16
Most recently, a clinical trial combining VPA, radiation, and chemotherapy for children with high-grade gliomas reported that three times daily administrations, to maintain plasma concentrations of 75–100 µg/ml, was well tolerated and histone hyperacetylation in peripheral blood mononuclear cells (PBMCs) was observed in half of the patients at steady state. 18
In a phase-I/II trial, 44 patients with solid tumors received escalating doses of VPA in combination with a fixed dose of epirubicin alone, or in combination with 5-fluorourcail (5-FU), epirubicin, and cyclophosphamide (FEC100), and the MTD for VPA was 140 mg/kg/day, with nine patients achieving a partial response. During the second part of the study, a disease-specific cohort of 15 breast cancer patients were treated with 120 mg/kg/day VPA plus the FEC100 regimen, with 9 out of 14 patients responding to therapy. Overall, somnolence was the most noted adverse effect related to VPA and the acetylation levels measured in PBMCs correlated with VPA serum levels and could be linked to baseline HDAC2, but not HDAC6, expression. 17
VPA safety and cardiac toxicity
Common adverse effects associated with HDACi include thrombocytopenia, neutropenia, diarrhea, nausea, vomiting, and fatigue. Most toxicities are not class-specific and have been observed with all HDACi, with the exception of VPA, where somnolence appears to be dose-limiting, rather than fatigue. Indeed, in contrast to other HDACi, VPA has a good tolerability and safety profile with predominantly neurovestibular DLT observed (dizziness, confusion, and hearing loss).12,13 Several additional mild and transient side effects were described for VPA, but most of them were related with its chronic use. 12 In detail, weight gain, changes in serum triglycerides, cholesterol, and fast glucose were described, as well as some dermatological effects such as stomatitis, cutaneous leukoclastic vasculitis, and psoriasis-like eruption. Owing to its direct neurological action, some rare neurological side effects were also reported, including encephalopathy, VPA-induced parkinsonism, and hyperammonia in the absence of liver failure. Hepatotoxicity has been also reported, particularly in young children, and in the presence of hepatic disorders. Finally, one study reported the increased risk of aplastic anemia after the use of VPA, but opposite evidences reported VPA as a potent activator of erythropoiesis in epileptic patients.
Extensive studies have been performed to determine whether HDACi are associated with cardiac toxicities. To date, there is little conclusive evidence to determine whether some, or all HDACi, cause electro-cardiac changes, including QT-prolongation.
In the two phase-II clinical trials submitted for the approval of the HDACi vorinostat, a single event of myocardial infarction and three sudden deaths were reported, 6 without clinically significant changes in left ventricular ejection fraction. Cardiac toxicity was reported as DLT in a phase- I study with the HDACi depsipeptide, 19 reporting one episode of grade 4 atrial fibrillation, with reversible ST/T changes and mild reversible dysrhythmias, observed on the post-treatment ECG, and three episodes of asymptomatic dysrhythmias. In another phase-I trial, 20 minimal cardiac effects were observed with depsipeptide treatment: one patient developed a transient left bundle branch block 22 h after completion of the first dose, that resolved spontaneously the following day and did not recur with subsequent depsipeptide treatments; another patient developed an isolated elevated troponin level with the first dose of depsipeptide treatment that was transient and not associated with any other serologic, EKG, or echocardiographic evidence of myocardial ischemia or infarction; no clinically significant changes in left ventricular ejection fraction were reported. A phase-II study with depsipeptide in neuroendocrine tumors was prematurely terminated due to an unexpected high number of serious cardiac adverse events: one sudden death attributed to possible fatal ventricular arrhythmia occurred within 24 h after the fifth dose of depsipeptide; two case of asymptomatic grade 2 ventricular tachycardia; and five case of prolonged QTc; all probably related to depsipeptide. 21 Finally, in a phase-I trial of VPA in combination with epirubicin, grade 2 QTc prolongations were seen in eight patients (18%), and grade 3 QTc prolongations in two patients (5%). These events occurred predominantly on day 1 of VPA treatment, not day 3, of the cycle. QTc prolongations were associated with serum potassium levels less than 4.0 mmol/l and were resolved in all patients with appropriate potassium and magnesium supplementation. 12
Rationale for the combination of VPA with fluoropyrimidines, oxaliplatin, and bevacizumab
Our group has demonstrated the synergistic antitumor activity of HDACi in combination with a large number of structurally different anticancer agents, including 5-FU, capecitabine, and DNA-damaging compounds such as platinum-based agents.22–25 In details, we have previously demonstrated that the HDACi are able to modulate the levels of two critical enzymes in the metabolism of fluoropyrimidines, such thymidylate synthase (TS), the target of 5-FU, and thymidine phosphorylase (TP), the critical enzyme converting, within tumor cells, capecitabine metabolite into the active drug 5-FU. 24 In details, we showed that the HDACi vorinostat in combination with capecitabine produces a synergistic antitumor effects by up-regulating, in vitro and in vivo, in CRC cells but not in ex vivo treated peripheral blood lymphocytes, the mRNA and protein expression of TP and by downregulating the expression of TS. 24
Recently, we also demonstrated that several HDACi, including VPA, down-regulate TS and up-regulate TP mRNA and protein expression in breast cancer cell lines. VPA modulates TP at a transcriptional level and this alteration involves mainly HDAC3 isoform. 26 Moreover, by using a stable TP-knockdown cell model, we demonstrated that TP has a critical role both in vitro and in vivo synergistic antitumor effects of HDACi in combination with capecitabine. 26 Notably, washout experiments showed that the induction of TP, mediated by VPA treatment, is still evident 24 h after the drug removal, suggesting the feasibility of a sequential schedule of combination treatment. 26
Finally, we recently discovered for the first time that VPA/fluoropyrimidines combination further synergizes with RT in CRC models, 27 also confirming the VPA-induced modulation of both TS and TP protein levels in the presence of RT. Interestingly, TP protein induction is achieved also at low doses of VPA (0.5–1 mM), corresponding to a plasma level between 50 and 100 μg/ml, easily reached in patients with normal anticonvulsant doses and included in the clinical limits of tolerance. 27 Interestingly, although at these doses VPA did not induce growth inhibition as single agents, a significant synergistic antitumor effect was still demonstrated in combination with fluoropyrimidines and RT, suggesting a specific mechanism of interaction as well as the feasibility to translate this approach in a clinical study. 27 Mechanistically, the observed synergism between VPA, fluoropyrimidines and RT, involves increased DNA damage, DNA repair disruption and induction of apoptosis. 27 Indeed HDACs have recently been found to participate in the DNA damage response and their down-regulation has been associated with impaired DNA repair. 28
On these basis, we are currently exploring VPA at antiepileptic dosage, in combination with capecitabine, during preoperative radiotherapy in locally advanced rectal cancers patients (V-ShoRT trial). 29 Data from the completed phase I study showed the feasibility of VPA in combination with chemo-radiotherapy.
Cancer stem cells (CSCs) with the ability to drive tumor growth have been identified in human cancers, including CRC, and were associated with chemoresistance and minimal residual disease. 30 Interestingly, unpublished results from our group demonstrated that VPA induces cellular differentiation and sensitization of CRC CSCs to oxaliplatin. Notably, preclinical data also suggest that RAS-mutated cancer cells are more prone to DNA damage and apoptosis induced by HDACi. 31
Preclinical and clinical data suggest that HDACi, by modulating VEGF expression, may prevent/reverse anti-angiogenic resistance and potentiate the efficacy of anti-angiogenic drugs. 32 Several clinical trials explored the combination approach of an HDACi plus bevacizumab, alone or plus chemotherapy, particularly in glioblastoma patients but also in other solid tumors.33–43 VPA has also been shown to inhibit endothelial function and angiogenesis, in vitro as well as in vivo, and to alter angiogenesis in human cancer models.44–52 The combination of bevacizumab and VPA is safe in patients with advanced malignancies, with clinical activity demonstrated in CRC, gastroesophageal junction and prostate cancer. 53
Rationale for the biologic pharmacodynamic and pharmacokinetic studies
There is an unmet need for pharmacodynamics and predictive biomarkers in cancer therapy. The identification of predictive biomarkers, of toxicity and efficacy, as well as those for therapeutic drug monitoring leading to dosage optimization, could improve patients quality of life (QoL), treatment costs, and the implementation of the treatment approach evaluated within this trial into clinical practice.
Metabolomics evaluation by nuclear magnetic resonance (NMR) is a novel potent and affordable approach to select novel biomarkers. 53 We will evaluate in peripheral blood metabolomic profiling at baseline and during treatment by NMR, to early distinguish patients for whom the treatment is effectively beneficial, based on our preliminary experience in CRC patients. In details, recently, we evaluated by NMR spectroscopy the metabolomic profiling on sera from 20 healthy controls and 22 mCRC patients within OBELICS trial, the latter subdivided on the basis of outcome, demonstrating that good versus bad responders and mCRC versus controls, grouped in separate clusters.
We will evaluate molecular determinants on circulating free DNA (cfDNA) and on tumor tissues (when available), at baseline and during treatment (i.e. RAS), by next generation sequencing (NGS), since we have previously reported that liquid biopsy enables real time monitoring of molecular alterations in mCRC during treatment. 54
We will also evaluate several tissue biomarkers comparing normal mucosa with tumor at baseline (possibly within the diagnostic biopsy), and on tumor tissues (when available, either primary tumor and/or resected metastases) during treatment, by immunohistochemistry (IHC) and real-time polymerase chain reaction (PCR), to confirm our preclinical published and preliminary data. In details, TS and TP expression will be evaluated. A specific panel of CSC (i.e. LGR5) and differentiation (i.e. CK20) markers, selected preclinically, will be also studied to confirm the mechanistic insight (preliminary data). HDAC isoforms (at baseline) and histones and proteins acetylation (H&P-Ac), will be studied to be correlated with VPA effect. Angiogenesis markers (i.e. VEGF) and the expression of DNA repair genes (i.e. RAD51, XRCC1) will be also evaluated.
Over the past decade several reports demonstrated that circulating endothelial cells (CECs) levels are increased in the peripheral blood of cancer patients at diagnosis, and chemotherapy can reduce the amounts of mature viable CECs, determining the return to normal values in patients undergoing complete remission. We participated in a multicenter study to align CEC phenotypes and counts in peripheral blood, establishing a polychromatic flow cytometry procedure 55 that might be adopted as standardized method and provided a reference physiological baseline range for the healthy population, useful for CEC monitoring in endothelial dysfunctions, including cancer-related angiogenesis. We will evaluate CEC counts on peripheral blood at baseline and at an early time point (day 10–15) selected by our previous experience, 56 as a surrogate marker of tumor angiogenesis and pharmacodynamics readouts of bevacizumab activity; we will also test a large panel of circulating angiogenic factors (i.e. angiopoietin, sTIE, Ang-2, VEGF isoforms, PDGF, PlGF, sVEGF-R-1 and -2 isoforms) and circulating cytokines profiling, by a multiplex biometric enzyme-linked immunosorbent assay (ELISA)-based immunoassay in the plasma, to be correlated with bevacizumab pharmacokinetics and pharmacodynamics data as well as to treatment efficacy and toxicity.
The pharmacokinetics of monoclonal antibodies (MoAbs) are complex and different from other anticancer agents, but their inter-individual variability on pharmacokinetic processes is similar to that observed for small-molecule anticancer drugs. MoAbs are dosed by body weight or at fixed doses but high variability in MoAb exposure has been observed after their administration at labeled dose, supporting the need to individualize dosing to account for variability and ensure efficacy. Therapeutic drug monitoring studies for MoAbs are limited and a strong exposure–response relationship have been described in solid tumors only for trastuzumab and cetuximab. No such studies have been performed for bevacizumab. Indeed, bevacizumab has been approved in different cancer disease and with different dosages with minimal clinical benefit reported in terms of outcome. Formal clinical evaluation of the optimal dosing of bevacizumab will increase knowledge about the exposure–response/toxicity relationship and could be of importance to determine whether this treatment strategy can be individualized to further improve its benefit. The availability of a reproducible, simple, and rapid method to quantify plasma or serum bevacizumab concentration could allow, within the trial, the determination of individual pharmacokinetic parameters and the proposal of new doses adjusted to the individual needs of each patient (i.e. pharmacokinetic target to efficacy and toxicity), considering that chronological age is not as important as physiologic age/frailty. We will correlate bevacizumab pharmacokinetics to efficacy and toxicity as well as to VEGF concentrations and other angiogenesis biomarkers such as CEC.
Normally, in humans, 80–90% of standard administered doses of 5-FU are rapidly metabolized in the liver to 5,6-dihydro-5-fluorouracil by dihydropyrimidine dehydrogenase, the key enzyme of 5-FU catabolism, and only a small fraction can exert its action. 57 Therefore, the drug is administrated at high doses to maintain the therapeutic concentration. Response to 5-FU treatment is highly variable among patients, of whom 10–30% experience severe to life-threatening adverse effects at standard doses, such as mouth sores, swallowing difficulties, diarrhea, nausea and vomiting, and low white blood cells count. 57 Since toxicity and response rates clearly correlate to 5-FU plasma levels in different administration schedules, drug levels measurement in plasma can help to monitor 5-FU dose and to develop an optimum procedure for its administration. We recently developed and validated a sensitive and specific liquid chromatography (LC)-UV method for the analysis of 5-FU in human plasma. 57 In addition, 5-FU plasma concentrations will be correlated with efficacy and toxicity.
Histone acetylation in tumor samples and in PBMC correlated in several studies with VPA serum levels and were also further linked to baseline expression of some HDAC isoforms (i.e. HDAC2 but not HDAC6). Within our ongoing phase-I/II V-ShoRT study, we have optimized a protocol to evaluate H&P-Ac in peripheral blood leucocytes (PBLs) of recruited patients, by flow cytometry, as pharmacodynamics/predictive specific marker of VPA HDAC inhibitory activity. H&P-Ac on PBL at baseline and during treatment could then be evaluated as pharmacodynamics readouts of VPA activity; VPA serum level during treatment to be correlated with PBL H&P-Ac, is an additional markers of VPA activity.
Moreover, polymorphisms of several genes may affect activity and toxicity of VPA, bevacizumab, or chemotherapy, by regulating either dosing of the drug itself or the blood levels of the target (i.e. UDP-glucuronosyltransferases, VEGFA and VEGFR1, DPD, TS, XRCC1, GSTP1, RAD51, and XRCC3), and can be easily evaluated on genomic DNA isolated from whole blood.
Finally, mounting evidence indicates an enhanced lymphocytic reaction as an informative prognostic indicator in CRC; 58 moreover the VEGF suppressive role on immune cells and the improvement in tumor immune responses combining anti-angiogenic drugs and checkpoint therapies was recently reported; 59 furthermore, an immunomodulatory effect of HDACi was also reported. 60 On this basis, we will evaluate by IHC the prognostic and predictive value of immunoscore (density of tumor infiltrating CD3 and CD8) and of PDL-1, on resected primary tumor and metastases and the difference between the two when possible. We will also evaluate myeloid derived suppressor cells (MDSCs) and T-cell subpopulations in peripheral blood at baseline and during treatment, as readout of immune response.
Methods/design
Revolution is a prospective, randomized, open-label, two-arm (1:1), multicenter phase-II trial evaluating the potential of VPA in combination with bevacizumab and oxaliplatin/fluoropyrimidine standard first-line regimens in RAS mutated mCRC patients. Correlative studies aim to identify predictors of treatment response/resistance and to add new insight in the mechanism of interaction between HDACi and anti-angiogenic plus chemotherapy treatment.
Objectives
The primary objective is to test whether the combination of VPA with bevacizumab and oxaliplatin/fluoropyrimidine regimens (mFOLFOX6/mOXXEL) can prolong progression-free survival (PFS) as compared with bevacizumab and oxaliplatin/fluoropyrimidine regimens alone, as first-line treatment in patients with mCRC with mutation of RAS. Secondary objectives are to evaluate the impact of the VPA combination on objective response rate (ORR), metastases resection rate, overall survival (OS), PFS according to independent centralized review (PFSCR), toxicity, and quality of life. Exploratory secondary objective is the evaluation of the prognostic and predictive value of several biomarkers on blood as well as tumor tissue samples (primary tumors and resected metastases). In details, on blood samples several biomarkers will be analyzed: (a) polymorphisms of several genes that may affect activity and toxicity of VPA, bevacizumab or chemotherapy, on DNA from whole blood; (b) mutations on cfDNA; (c) CECs counts; (d) MDSC and T-cell subpopulations; (e) H&P-Ac in PBL; (f) levels of drugs (5-FU, bevacizumab, and VPA); (g) circulating cytokines and chemokines; (h) metabolomic profiling. On tissue samples a comparative baseline DNA mutational status versus cfDNA (i.e. RAS), will be performed. Moreover, the immunoscore and PD-L1 expression, as well as mechanistic-based pharmacokinetic/pharmacodynamic tissue biomarkers will be evaluated (TS and TP, LGR5, HDAC isoforms, H&P-Ac VEGF, RAD51, and XRCC1).
Study design
Revolution is a randomized two arm (1:1) phase-II trial comparing in metastatic RAS-mutated CRC patients standard bevacizumab+oxaliplatin/fluoropyrimidine regimens (mFOLFOX-6/mOXXEL) or the combination of VPA with the same regimens (Figure 1). Oxaliplatin regimen (mFOLFOX/mOXXEL) is chosen according to local clinical practice at the beginning of the study.

Study design.
The study is planned as a randomized phase-II trial with PFS as the primary end point. Study sample size is defined accordingly to the expected median PFS of 9 months in the standard arm [pooled analysis of five trials (FIRE-1, FIRE-3, AIOKRK0207, AIOKRK0604, RO91) by AIO colorectal cancer study group], 1 and assuming a median PFS of 12 months in the experimental arm, corresponding to a HR = 0.75. The hypothesis is that the addition of VPA to chemotherapy and bevacizumab regimens can improve PFS by 3 months, with one-sided alfa of 0.20, a beta error of 0.20, and a power of 0.80. With the above parameters we plan to accrue 200 patients and we need to observe 137 events for the final analysis. No interim analysis is planned.
Patient selection criteria
Inclusion criteria
Patients are eligible if ⩾18 years, with histologically confirmed diagnosis of unresectable RAS-mutated mCRC, have at least one measurable target lesion (according to RECIST 1.1); have an ECOG performance status of 0–1 at study entry and a life expectancy >3 months. Moreover, at least 28 days should elapse from a surgical procedure or from performing a biopsy and they have to consent to use effective contraception if the risk of conception exists. Signed written informed consent is needed.
Exclusion criteria
Patients are excluded if they have RAS wild-type mCRC; have received previous chemotherapy for metastatic disease; or previous adjuvant chemotherapy if ended <6 months before relapse or <24 months if included oxaliplatin; or radiotherapy to any site for any reason within 28 days prior to randomization (palliative radiotherapy to bone lesions is allowed if ⩾14 days before randomization); or prior treatment with an HDACi or compounds with HDACi-like activity, such as VPA. Patient are also excluded if they have known or suspected brain metastases (determined exclusively in the presence of at least one clinical symptom); regularly use non-steroidal anti-inflammatory drugs (NSAIDs) or aspirin (more than 325 mg/day) or anticoagulants at therapeutic dose; have bleeding diathesis or coagulopathy; have inadequate bone marrow, liver or renal function (neutrophils < 2000/mm3 or platelets < 100,000/mm3 or hemoglobin <9 gr/dl; creatinine levels of >1.5 time the upper normal limit (UNL); GOT and/or GPT > 2.5 time the UNL and/or bilirubin >1.5 time the UNL in absence of liver metastasis; GOT and/or GPT > 5 time the UNL and/or bilirubin >3 time the UNL in presence of liver metastasis; had any other malignancy other than non-melanomatous skin cancer, or carcinoma in situ of the cervix or surgical-resected prostate cancer with normal PSA within 5 years. Patients HIV positive; with DPD deficiency; with inadequately controlled hypertension, clinical significant cardiovascular disease, long QT syndrome; with active or uncontrolled infection, or any other serious uncontrolled medical disorder that in the opinion of the investigator would impair the ability to receive protocol therapy, are also excluded. Pregnancy or breast-feeding women are also excluded.
Treatment plan
In the standard arm bevacizumab is administered at dose of 5 mg/kg as 20–30 minute intravenous (i.v.) infusion before oxaliplatin on day 1 of each cycle of mFOLFOX-6 (oxaliplatin 85 mg/m2 as 2–3 h i.v. infusion on day 1 followed by levo-folinic acid 200 mg/m2 as 1–2 h i.v. infusion followed by i.v. bolus. 5-FU 400 mg/m2, and a 46-hour i.v. infusion of 5-FU 2400 mg/m2 every 2 weeks for 12 cycles); or mOXXEL regimen (oxaliplatin 85 mg/m2 as 2–3 h i.v. infusion on day 1 plus oral capecitabine 1000 mg/m2 twice daily on days 1–10, every 2 weeks for 12 cycles).
In the experimental arm VPA will not have a predefined dose but a titration strategy will be applied in each patient to achieve a serum concentration included between 50 and 100 μg/ml. This target serum level range is considered to produces the desired synergistic effect with chemotherapy and also represents the recommended values for the treatment of epilepsy. VPA will be administered orally starting at day −14 with 1 × 500 mg slow releasing tablet at evening. Thereafter, the dose will be increased accordingly to the scheme in Table 1, also using 300 mg tablets.
Valproic acid titration scheme.
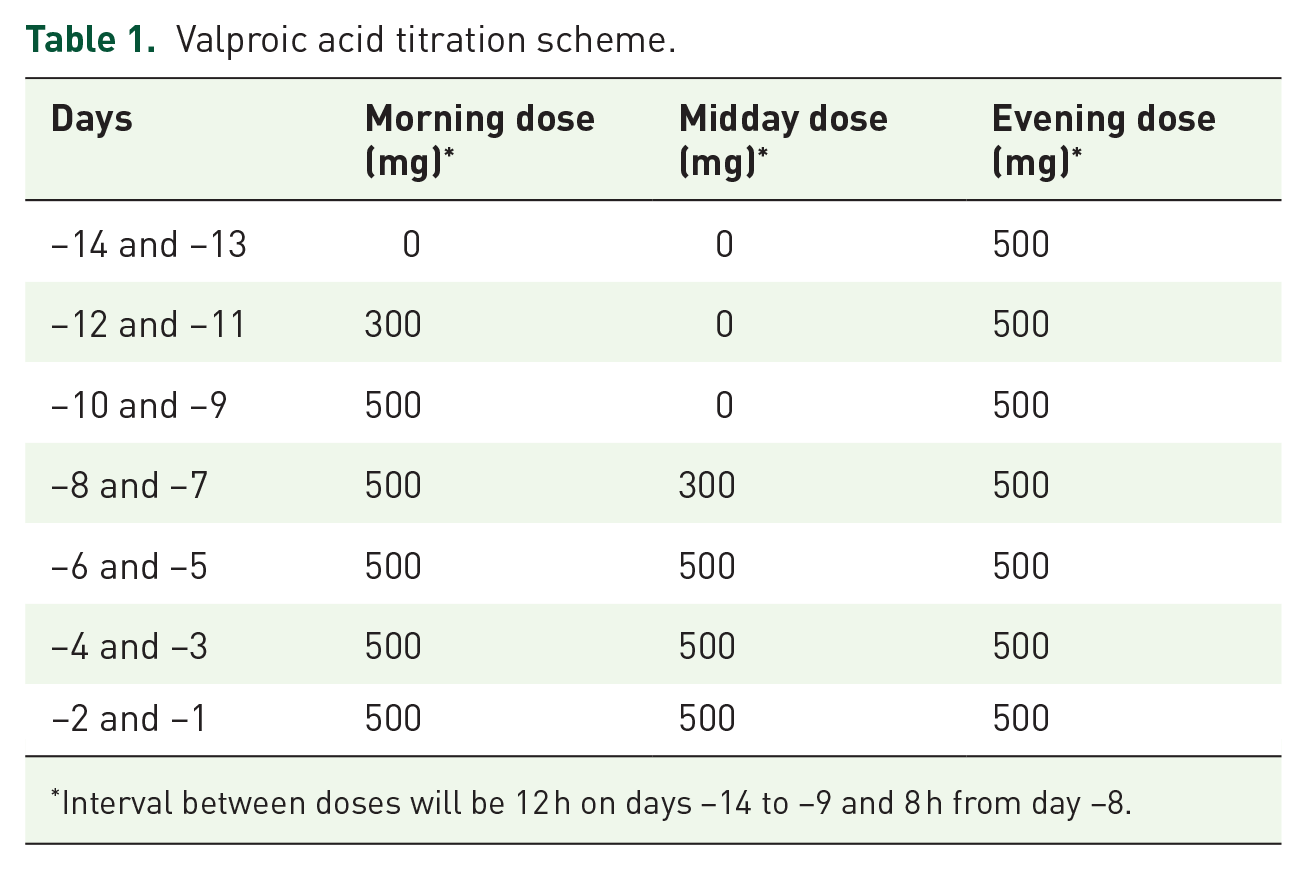
Interval between doses will be 12 h on days −14 to −9 and 8 h from day −8.
In the morning of day −4, serum level of VPA will be checked by drawing a blood sample within 2 h after taking the morning dose using a commercially available valproate test and will be adjusted depending on the reached steady level. If VPA level is <50 µg/ml, the VPA dose will be increased by 300 mg/day (adding one tablet at the evening dose) until the check of VPA level planned on day 1. If necessary, another 300 mg tablet can be added, at the midday dose will be achieved.
The converse will be done with VPA levels >100 µg/ml, with a reduction in total daily dose by 200 mg (this will be done by changing the morning 500 mg morning tablet with a 300 mg tablet). VPA level will be then checked again on day 1 and thereafter weekly, with the goal to keep target serum level between 50 and 100 µg/ml.
Patients who are progression free after 12 cycles (24 weeks) of treatment continue maintenance with fluoropyrimidine/bevacizumab in standard arm or same regimen plus VPA daily in experimental arm, until disease progression or unacceptable toxicity.
Surgery may be carried out in case appropriate tumor reduction is evident at response evaluation. Resectability has to be evaluated by a multidisciplinary review team and the decision regarding post-surgery chemotherapy is at the discretion of the investigators, according to the policy commonly adopted by their institution in clinical practice.
In case of grade ⩾ 3 of hematologic or non-hematologic toxicities, a 25% fluoropyrimidines dose reduction will be applied in subsequent cycles (except for alopecia); at the second appearance of these side effects a dose reduction of 50% of fluoropyrimidines may be applied. If one of these events occurs again, despite reducing the dose of fluoropyrimidines to 50% of the initial dose, a 25% reduction of the oxaliplatin dose will be applied. If grade 3 neurotoxicity with a duration less than 15 days occurs, the oxaliplatin dose will be reduced to 75% of the initial dose at the next cycle. If the event occurs again, a further dose reduction of the oxaliplatin at 50% of the initial dose will be performed. If the event will occur again, or if a grade 3 neurotoxicity with a duration >14 days occurs, or if a grade 4 neurotoxicity (any duration) appears, treatment with oxaliplatin will be permanently discontinued. If one of these events should occur despite all the described dose reductions, chemotherapy treatment will be definitively stopped, whereas bevacizumab alone or bevacizumab plus VPA may continue, as assigned previously.
No dose reduction of bevacizumab is planned. In case of hypertension grade > 3, proteinuria grade > 3 (nephrotic syndrome), thrombosis/embolism grade > 3, hemorrhage grade > 3, or grade > 3 left ventricular dysfunction (CHF), bevacizumab will be permanently discontinued. An antihypertensive treatment should be undertaken in case of hypertension grade ⩾ 2 (recurrent or persistent) or in the case of a symptomatic increase >20 mmHg (diastolic) or in the case of a pressure >150/100 mmHg if this was initially within the limits. The administration of angiotensin-converting enzyme (ACE) inhibitors and/or calcium antagonists is recommended, at the indicated standard doses. In case of unforeseen and urgent surgery, bevacizumab should be suspended as soon as possible and all the precautions should be taken.
In case of grade 2 somnolence or fatigue the VPA dose will be reduced by −200 mg/day steps up to reaching grade ⩽ 1 independently of the actual serum level. In case of grade ⩾ 3 somnolence or fatigue VPA will be definitely suspended. In case of asymptomatic QTc prolongation development (QTc > 500 ms, or QT prolongation >60 ms, but not associated with symptoms), VPA has to be suspended. Electrolytes and concomitant medications have to be checked and corrected. ECG has to be repeated after 24 h. If the event is resolved, treatment with VPA can be resumed but the dose will be reduced by −200 mg/day; in contrast, if QT prolongation is confirmed VPA has to be interrupted.61,62 In case of symptomatic QTc prolongation development (QTc > 500 ms or QT prolongation >60 ms, and is associated with symptoms suggestive of a ventricular tachyarrhythmia), VPA has to be interrupted.
Study procedures
Assessment and procedures, including those for exploratory objectives (see below) are illustrated in Figure 2.

Study procedures.
Response evaluation
Response evaluation is planned at the 12th and 24th week from randomization and thereafter every 12 weeks, until disease progression, by repeating CT scan of the chest, abdomen, and pelvis; CEA, CA 19.9; and any test resulting positive at baseline. Responses will be codified by the investigator, according to the RECIST (v 1.1). CT (or other pertinent) scans performed at baseline and during follow-up will be collected and reviewed also by an independent central panel of radiologists.
Toxicity assessment
Treatment toxicity will be evaluated at each study visit and adverse events will be reported according to the Common Terminology Criteria for Adverse Events of the National Cancer Institute (CTCAE-NCI) version 5.0. A Clinical examination before each cycle, measurement of blood pressure before of each administration of bevacizumab, a complete blood count and biochemistry at day 1 every treatment cycle. In the standard arm an ECG at 12th and 24th weeks from randomization and thereafter every 12 weeks will be performed. In the arm with VPA ECG will also be performed on days −11 and −4, and in patients with QT prolongation it will be repeated after 24 h. In both arms ECG and any other exam will be performed whenever considered clinically indicated by the investigator for the evaluation of adverse events. All serious adverse events (SAEs) occurring during treatment or until the last dose of study treatment, must be recorded and reported immediately and not exceeding 24 h following knowledge of the SAE.
Biomarkers
Analysis on biomarkers is exploratory by nature and will be performed in the majority of cases retrospectively after the main study analysis is completed. Since the identification of new markers correlating with disease activity and the efficacy or safety of treatment are rapidly evolving, the definitive list of analyses remains to be determined; however, several tests are planned and are described in the following.
Blood samples
Peripheral blood samples for exploratory biomarker studies are collected at baseline and at several time points during treatment (Figure 2). Further blood samples are collected at progression and, when patients undergo resection (primary and/or metastases), before and after surgery. Samples are aliquoted and stored at −80°C and analyzed as indicated in the following, with the exception of circulating cells that are processed and analyzed promptly.
Polymorphisms in genes that may affect activity and toxicity of the drugs (i.e. UDP-glucuronosyltransferases, VEGFA and VEGFR1, DPD, TS, XRCC1, GSTP1, RAD51, and XRCC3) will be analyzed by quantitative reverse transcriptase (qRT)-PCR using specific TaqMan probes.
cfDNA mutations will be evaluated by a dedicated developed panel, the SiRe panel, targeting all relevant clinical mutation KRAS, NRAS, BRAF, EGFR, cKIT, and PDGFR genes with an ultra-deep NGS approach (coverage per amplicon > 2000×). cfDNA mutational profiling will be automatically analyzed by a dedicated pipeline (IonReporter suite, Life Technologies) and also visually inspected by an expert molecular pathologist. Variants will be automatically annotated using variant caller plug in (v.5.0.2.1) setting specific panel optimized parameters. In particular, for cfDNA mutational analysis, only variants with 5× allele coverage and a quality score 20, within an amplicon covered at least 1000× will be called, and the frequency of each mutant allele will be recorded. Bioinformatics analysis of NGS data will be obtained and compared in the different time points.63,64
CEC are evaluated, after red cell lyses within 24h from collection of sample stored at 4°C by flow cytometry as described by Lanuti et al. 55 MDSCs and immune cell subtypes expression and lymphocyte activation are evaluated by multicolor cytofluorometric approach, taking advantage of two eight-color version panels, one exploring multiple immune cell subsets expression/activation (CD25, CD8, CD45RA, CD127, CD28, CD3, CD39, CD4) and one for MDSC identification (HLA-DR; CD11b, CD15, CD124, Lin, CD33, CD14, CD45). Histones and proteins acetylation will be evaluated by protocols developed in our lab by modifying those reported previously.65,66
Blood levels of drugs are measured as follows: VPA serum levels are measured by commercial test (only in patients assuming the drug); 5FU serum levels are measured by specific LC-UV method as previously described by Russo et al. 57 ; bevacizumab plasma concentration are measured on plasma by a LISA TRACKER Duo bevacizumab kit based on ELISA methodology (Theradiag, https://www.theradiag.com/en/theranostic-2/therapeutic-drug-monitoring-biologics/).
Circulating cytokines and chemokines will be evaluated by a multiplex biometric ELISA-based immunoassay in the plasma, accordingly to the manufacturer’s instructions (Bio-plex, Bio-Rad Lab., Inc., USA) as described by Capone et al. 67
Metabolomic profiling will be evaluated by NMR spectrometer (600 MHz). 1H and 2D spectra on sera samples will be acquired by 600-MHz Bruker Avance DRX NMR spectrometer with a TSI probe as reported previously by Sorice et al. 68 The metabolite assignments will be based on the comparison of chemical shifts and spin-spin couplings with reference spectra and tables present in the SBASE-1-1-1 database by AMIX package (Bruker, Biospin, Germany), the human metabolome database (HMDB), and the biological magnetic resonance database (BMRB). To compare the spectra obtained for the different patient groups, we will apply some statistical analysis such as principal component analysis, orthogonal partial least squares discriminant analysis (OPLS-DA), clustering, fold change analysis, and T-test. Identification of statistically differentially expressed metabolites between different time points will be performed by S-Plot and VIP plot. Moreover, a functional and pathway analysis will be done by different bioinformatic tools (STRING, DAVID, and Ingenuity pathway analysis).
Tissue samples
Tumor tissue samples are collected at baseline as formalin-fixed, paraffin-embedded (FFPE) tissue and/or frozen tissue from biopsies and surgical samples in resected patients, to assess exploratory biomarkers and also to compare expression levels of tumor tissue versus normal mucosa or baseline tumor tissues versus resected tumor/metastases after treatment. Comparative baseline DNA mutational status analysis versus cfDNA will be performed as described previously by NGS using SiRe panel. Moreover, DNAs derived tissues will be also tested by NGS using a 50-gene HotSpot Cancer panel (Life Technologies) to better define the additional mutational status in other cancer related genes. For the samples with a percentage of neoplastic cells <50%, a manual micro-dissection of the specimen will be performed, if possible. Genomic DNA will be extracted from FFPE samples using the QIAGEN DNA Mini Kit following the manufacturer instruction.
The following markers will be also measured on tumor and normal samples, including analysis of protein levels, and/or gene expression/amplification related to CRC biology and other exploratory markers related to the specific mechanism of action of the drugs administered to the patients (fluoropyrimidines, oxaliplatin, bevacizumab, VPA). Analysis of mRNA and protein expression are performed for crucial enzyme in the preclinical synergism observed between HDACi and fluoropyrimidines (i.e. TP, TS); specific markers of CSC (i.e. LGR5), differentiation (i.e. CK20) and angiogenesis (i.e. VEGF); DNA repair genes (i.e. RAD51, XRCC1); HDAC isoforms. RNA from tissue samples will be obtained using the RNA extraction kit (Qiagen/SA Biosciences) following manufacturer instructions. The expression of the indicated gene will be evaluated by real-time PCR with the specific primers and probes (TaqMan, Applied Biosystem). Proteins expression of the indicated genes as well as of histones and proteins acetylation will be evaluated by IHC on FFPE specimens.
Immunoscore as well the expression of PD-L1 on both tumor and immune cells on paraffin embedded tissue samples (FFPE; quantifying simultaneously CD8+PD-L1 for expression, density, proximity), will be assessed in a multiplexed standardized IHC assays by high-resolution image automated quantification system.
All the biomarkers described previously will be correlated to the clinical outcome to define the predictive and/or prognostic role of the alterations detected.
QoL assessment
QoL will be assessed in patients enrolled in the study by the EORTC QLQ-C30, version 3.0 69 and the EORTC QLQ-CR29 questionnaires that must be compiled by the patients before any study procedure but within 2 weeks prior to randomization (baseline) and at weeks 12 and 24 during treatment, and subsequently every 3 months in both arms. For patients who progress during treatment, QoL assessment stops at the last assessment before progression.
Statistical analysis
All analyses will be performed according to the intention-to-treat (ITT) strategy. All subjects who received at least one dose of the assigned study treatment will be included in compliance and safety analyses. No “per-protocol analysis” will be performed.
PFS is defined as the time elapsed from the date of randomization to the date of progression, as defined by investigators, or the date of death, whichever comes first. Patients lost to follow up or alive at the end of the study will be censored on the date of the last tumor assessment. PFS curves will be described according to the Kaplan–Meier product-limit method. Data will be also presented as median, 95% confidence interval of the median, and point estimates at 6-month intervals. Treatment groups will be compared by unstratified log rank test. The hazard ratio (HR) of PFS will be estimated by a Cox model, with stratification factors as covariates (for this purpose, centers will be categorized into three categories according the volume of patient enrolled). Two-sided 95% confidence interval of the HR will be provided. Heterogeneity of treatment effect across major subgroups will be described in a Forest plot.
OS is defined as the time elapsed from randomization to death due to any cause. Patients still alive at the end of the study will be censored on the date of the last information on vital status. OS curves will be described according to the Kaplan–Meier product-limit method. Log-rank test will be applied to test statistical significance of the differences.
As secondary analysis PFSCR is also measured, as the time elapsed from the date of randomization to the date of progression, as defined by independent blind central review, or the date of death, whichever comes first. PFSCR will be analyzed similarly as PFS in the primary analysis. However, if large differences were evident further analyses and any reasonable approach will be applied to understand the reason of the discrepancy.
ORR is defined as the ratio of patients who will experience a complete or partial response (according to RECIST v 1.1) out of those randomized with at least one target lesion at baseline. Response rates in the two arms will be described with their 95% confidence limits and will be compared with chi-squared test in a 2 × 2 contingency table (responders/non-responders × treatment arms).
The resection status and tumor regression will be evaluated by histopathological assessment of excised metastases together with the operation notes. Patients achieving a pathological tumor regression grade (TRG) 1 or 2 according to the Mandard scale, 70 will be defined as responders. Patients who will not undergo primary surgery because of progressive disease will be defined as non-responders. TRG rates will be compared with chi-squared test in a 2 × 2 contingency table (responders/non-responders × treatment arms). Metastatic resection status rates (R0/R1/R2), where R0 is no cancer cells seen microscopically, R1 is cancer cells present microscopically, and R2 is tumor tissue at naked eyes at the resection margin, will be described in the two arms, with their 95% confidence limits and will be compared with chi-squared test in a 2 × 2 contingency table [responders(R0)/non-responders × treatment arms].
For each patient and for each type of toxicity described according to CTCAE, the worst degree ever suffered during treatment will be used for the analysis. Two sets of statistical analyses will be performed to compare toxicity between the two arms. In the first set the whole pattern of toxicity (all grades) will be considered for each item; analysis will be done by a linear rank test. In the second set, toxicity will be defined as severe (grade 3 or higher) and not severe (including grades up to 2) and analysis will be performed by Fisher’s exact test.
EORTC questionnaires will be managed according to standard rules for their analysis reported in the EORTC manuals. 69
Owing to the small sample size, statistical analysis of biomarkers data will be conducted with the aim of hypothesis generation. First, a complete description of data from biological and pharmacogenomic studies will be done. For biomarkers that might change over time as a consequence of treatment, levels before and after treatment will be compared with appropriate statistical tests, based on the type of data. Serum levels of VPA throughout treatment will be described and compared between different acetylator phenotypes, with appropriate statistical tests. p values ⩽ 0.05 will be considered significant, and no adjustment is planned for multiple comparisons due to the exploratory nature of the analysis.
Discussion
Despite the evolution of treatments, prognosis of mCRC is still poor, mostly in RAS-mutated tumors, and resistance is a major challenge for treatment. Thus, particularly in RAS-mutated setting, intrinsic cancer biology factors suggest the need of novel combinatorial therapies. Our proposal will address such challenges by evaluating for the first time a novel therapeutic strategy based on the use of VPA, a safe and generic drug with HDACi activity, in combination with a consolidated treatment in this setting.
In detail, with the Revolution trial, by exploring the efficacy and safety of VPA in combination with bevacizumab and oxaliplatin/fluoropyrimidine regimens (mFOLFOX-6/mOXXEL), the standard first-line treatment in patients with RAS-mutated mCRC, we expect to validate a novel and affordable therapy in this poor prognosis mCRC group. Notably, national health systems, particularly the Italian SSN, are unlikely to support the explosion in costs of new oncology drugs for much longer. In light of this situation, optimization of consolidated anticancer drugs as well as mechanistic-based repurposing in cancer treatment of cheap and safe non-anticancer drugs already in clinical practice, such as VPA, represent an attractive strategy to offer more effective and affordable treatments to cancer patients.
We do not anticipate problems regarding the compliance of the patients to treatment considering that VPA is a very well-known and safe drug and that our intra-patient treatment approach should guarantee a good compliance, as also confirmed by preliminary data on the absence of adverse events experienced in the V-ShoRT trial. 71 Anyhow, we have planned, to ensure good compliance of the treatment, a pre-planned dosage reduction/treatment interruption policy in case of adverse events.
By evaluating potential predictive biomarkers of toxicity and efficacy as well as drugs pharmacokinetics we expect to improve dosage optimization by approaches that could be implemented into the clinical practice. Indeed, there is an unmet need for pharmacodynamics and predictive biomarkers in cancer therapy that we will try to address with a large program of correlative studies, including, among others, metabolomics evaluation by NMR, a novel potent and affordable approach that will be tested for the first time within a randomized clinical study in mCRC.
By predicting (non)-responders, we will also enable a personalized medicine approach, and suggest potential novel rational combinations. Furthermore, considering very limited experience of therapeutic drug monitoring, particularly with MoAbs such as bevacizumab, our study could increase knowledge about exposure–response/toxicity relationship to optimize therapy to individual needs of each patient.
Finally, whatever the result, Revolution trial, particularly the correlative studies, will also add new insights into the mechanism of interaction between HDACi, fluoropyrimidine, oxaliplatin, and bevacizumab.
Ethics and dissemination
Ethical aspects
The procedures set out in this study protocol are designed to ensure that the principles of the Good Clinical Practice guidelines of the International Conference on Harmonization (ICH) and the Declaration of Helsinki are respected in the conduct, evaluation, and documentation of this study. The study was approved by the Ethical Committee of the National Cancer Institute of Naples, Italy (Prot. 34/18). Patients provide written informed consent for participating in the study and for allowing the collection of tissue and blood samples.
Registration and data collection procedures
Patient registration, randomization, and data collection including radiologic examinations repository for independent assessment and tools for monitoring through e-queries, are centralized at the Clinical Trials Unit of the National Cancer Institute of Naples and are web-based (http://www.usc-intnapoli.net). Randomization is performed with a minimization procedure accounting for center, ECOG performance status (0 versus 1), site of primary disease (left versus right), and presence of synchronous versus metachronous metastasis. Biological analyses are centralized at the Experimental Pharmacology Unit of the NCI of Naples. The study started in May 2019. We planned to complete recruitment within 30 months and follow-up within further 10 months. Analysis as well as an end of the study are planned to be completed within 42 months from first patient recruitment. We planned five active participant centers.
Trial sponsorship
This is a non-profit investigator initiated trial (EudraCT: 2018-001414-15; ClinicalTrials.gov identifier: NCT04310176) supported by a peer-reviewed grant from the Italian Ministry Health (RF- 2016-02363314).
In this trial, bevacizumab, oxaliplatin and fluoropyrimidines are used on-label in accordance with approved indications, and does not increase costs for the National Health Care System. Valproic acid (Depakin Chrono®) will be bought by the promoter (National Cancer Institute of Naples).
The promoter will provide an insurance policy to cover possible damages caused to patients participating in the trial.
Footnotes
Author contributions
Conception and design: AA, MP, AB.
Development of methodology: AA, MP, ED, CR, FC, MR, FT, VG, AC, EC, NM, PM, LS, AD, FG, GR, ET, PA, AN, CV, RC, AL, AP, GB, PD, FI, FP, AB.
Writing, review, and/or revision of the manuscript: AA, MP, ED, AB.
Administrative, technical, or material support (i.e. reporting or organizing data, constructing databases): AA, MP, ED, VV, AB.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Italian Ministry of Health (RF-2016-02363314), POR CAMPANIA FESR 2014/2020 Progetto: Campania Onco-Terapie CUP: B61G18000470007, Fondazione Italiana per la Ricerca sul Cancro (FIRC)-AIRC supported with a triennial Fellowship to M.S. Roca (ID 21113).