
Abstract
Background
Dysbiosis commonly occurs in pancreatic cancer, but its specific characteristics and interactions with pancreatic cancer remain obscure.
Materials and methods
The 16S rRNA sequencing method was used to analyze multisite (oral and gut) microbiota characteristics of pancreatic cancer, chronic pancreatitis, and healthy controls. Differential analysis was used to identify the pancreatic cancer-associated genera and pathways. A random forest algorithm was adopted to establish the diagnostic models for pancreatic cancer.
Results
The chronic pancreatitis group exhibited the lowest microbial diversity, while no significant difference was found between the pancreatic cancer group and healthy controls group. Diagnostic models based on the characteristics of the oral (area under the curve (AUC) 0.916, 95% confidence interval (CI) 0.832-1) or gut (AUC 0.856; 95% CI 0.74, 0.972) microbiota effectively discriminate the pancreatic cancer samples in this study, suggesting saliva as a superior sample type in terms of detection efficiency and clinical compliance. Oral pathogenic genera (Granulicatella, Peptostreptococcus, Alloprevotella, Veillonella, etc.) and gut opportunistic genera (Prevotella, Bifidobacterium, Escherichia/Shigella, Peptostreptococcus, Actinomyces, etc.), were significantly enriched in pancreatic cancer. The 16S function prediction analysis revealed that inflammation, immune suppression, and barrier damage pathways were involved in the course of pancreatic cancer.
Conclusion
This study comprehensively described the microbiota characteristics of pancreatic cancer and suggested potential microbial markers as non-invasive tools for pancreatic cancer diagnosis.
Introduction
Pancreatic cancer (PC) is one of the most fatal malignancies with an insidious onset and grave prognosis. 1 Most patients manifest in the advanced stage when surgical resection offers minimal benefit. 2 Conventional therapy is ineffective, and it is notable that early detection could most likely improve the survival rate. 3 There were 460,000 new cases of PC and 430,000 deaths worldwide in 2018, and the incidence is predicted to increase in the coming decades. 4 Given that the management of PC is quite challenging, it is essential to improve research in the PC-related fields of pathogenesis, early detection, and treatment.
To date, the etiology of PC is currently unclear and is hypothesized to involve a combined effect of genetics and environment. Major risk factors include family heredity, cigarette smoking, chronic pancreatitis (CP), etc. 1 Notably, only 5–10% of patients have a familial basis, 5 whereas the genetic contribution in the other cases is unknown, suggesting the importance of environment in the etiology. The commensal microbiota, which has recently been recognized as a vital environmental factor, are associated with some gastrointestinal tumors, including PC. 6 Dysbiosis of commensal microbiota is commonly seen in patients with PC, but its specific characteristics remain uncertain due to conflicting conclusions among similar studies, which may result from small study size, uniform sample types, different regions, and experimental design limitations (lack of a benign disease group or matched controls).7–11 In view of the current status of relevant research, it is reasonable to scientifically conduct studies on the microbiota of Chinese patients with PC.
Here, we developed rigorous enrollment criteria and accordingly recruited three groups of participants including patients with pancreatic cancer, patients with CP, and healthy controls (HCs); we then instructed them to provide saliva and stool samples at the same time. We described and analyzed the oral and gut microbiota characteristics of the enrolled PC patients simultaneously. In addition, we also established diagnostic models based on oral and gut microbiome, to explore the potential of microbial markers for PC.
Materials and methods
Participant enrollment and sample collection
This study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of Changhai Hospital and Air Force Hospital of Eastern Theater Command.
There were three groups of participants in this study: PC patients, matched HCs, and CP patients. Patients with PC were recruited from inpatients of Changhai Hospital with confirmation by operation and histopathology. HCs were recruited from the healthy examination population of the Air Force Hospital, and the check-up results of these individuals were normal. CP patients were recruited from inpatients of Changhai Hospital and confirmed by clinical diagnosis. The inclusion criteria were operable and treatment-naïve PC or confirmed CP, and HCs were frequency matched for age, gender, race, and body mass index (BMI). Situations that affect commensal microbial composition (antibiotic/probiotics/proton pump inhibitor use within 4 weeks, anticancer treatment, infectious diseases, etc.) were rigorously excluded. Detailed enrollment criteria are described in the
Sample collection, process and storage were conducted with the uniform protocols. Participants were issued sterile containers and instructed to retain specimens normatively. Each participant provided a fresh fecal sample between 7:00 a.m. and 9:00 a.m., and an unstimulated saliva sample was collected. Fecal samples were divided into three 1-g aliquots, and saliva samples were divided into two 1.5 mL aliquots in a standard laboratory biosafety cabinet. Afterwards, they were labeled and stored at −80°C immediately. A total of 115 saliva/fecal sample pairs were prospectively collected, and 88 saliva samples (37 PC, 15 CP, and 36 HC) and 92 fecal samples (38 PC, 15 CP, and 39 HC) were finally enrolled.
DNA extraction and 16S rRNA sequencing
Salivary bacterial DNA was extracted using the QIAamp DNA Mini Kit (No. 51304, Qiagen, Hilden, Germany), and fecal bacterial DNA was extracted using the QIAamp Fast DNA Stool Mini Kit (No. 51604, Qiagen).
The V3-V4 16S regions were amplified by polymerase chain reaction and then sequenced as Ren, et al. 12 described. The sequencing data were classified according to specimen type, and cases were renamed as AS (saliva samples form PC patients), CS (saliva samples from matched HCs), YS (saliva samples from patients with CP), AF (fecal samples from PC patients), CF (fecal samples from matched HCs), and YF (fecal samples from patients with CP). All samples’ raw reads have been deposited in Sequence Read Archive (SRA) database under accession number PRJNA680233 (saliva samples) and PRJNA680467 (fecal samples).
Sequencing data clustering and operational taxonomic unit annotation
Assembled tags labeled with barcodes were then checked for length (restricted between 250 bp and 500 bp) and assessed for their average base quality (average Phred score of bases not less than Q20 with no greater than 3 ambiguous Ns). Eligible sequences were clustered into operational taxonomic units (OTUs) with > 97% identity using UPARSE 13 (http://drive5.com/uparse/), and chimeric reads were filtered by Usearch 14 (version 9.2). OTUs were assigned to taxonomy using the RDP database 15 (http://rdp.cme.mse.edu/).
Microbial diversity and functional analysis of 16S rRNA sequencing
The microbial diversity of samples was assessed in the form of alpha-diversity presented by the Chao1, Shannon index, observed species, and PD whole tree, which were calculated using the vegan package. 16
The significance of compositional difference among three groups was analyzed in the form of beta-diversity presented by principal coordinates analysis (PCoA) using R package (http://www.R-project.org/). The weighted and unweighted UniFrac distances were computed in a R phyloseq package.
The characterization of oral/gut microbiota was presented using the linear discriminant analysis (LDA) effect size (LEfSe) method 17 (http://huttenhower.sph.harvard.edu/lefse/) with the Kruskal-Wallis rank sum test (P < 0.05). The commensal functions were predicted with Phylogenic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), 18 and the differences among groups were analyzed using the LEfSe method as described above.
Genera biomarker identification and diagnostic model construction
Genera from the discovery set and validation set were mapped to generate discovery genus profiles and validation genus profiles, respectively. The Wilcoxon test (P < 0.05) was used to screen significant genus biomarkers for further analysis. An RF model (random Forest package) was evaluated by ten-fold cross-validation (CV) with default parameters except for “importance = TRUE” using the screened 21 genera profile of the discovery set (22 AF vs. 16 CF; 20 AS vs. 16 CS) as Ren, et al. 12 described. The error curve was gained by 10 trials of CV. The smallest CV error point was labeled the cutoff point, and its value was determined by the smallest error plus the SD at the corresponding point. Hence, the minimum number of genera was determined to be 10. Thus, the optimal set was established, and the receiver operating characteristic (ROC) curve was plotted (pROC package) to assess the established models. The area under the ROC curve (AUC) was computed to evaluate the ROC effect. The diagnostic efficiencies of gut genus biomarkers and oral genus biomarkers were evaluated based on the AUC value.
Statistical analysis
The differences among the three groups were compared with one-way analysis of variance. Continuous variables between both groups were compared by the Wilcoxon test; categorical variables were compared by Fisher's exact test; statistical analyses were conducted by SPSS 25.0 software (IBM Corporation, Chicago, IL, USA) (P < 0.05).
Results
Participant enrollment and demographics
Ultimately, there were 94 volunteers (40 patients with PC, 15 patients with CP, and 39 age-, gender-, and BMI-matched HCs) included in this study. Saliva and fecal samples were collected simultaneously. With rigorous exclusion processes, 180 valid sequencing results were ultimately obtained. RF models based on salivary or fecal microbiomes were constructed with sequencing data randomly divided into discovery sets (20 AS, 16 CS; 22 AF, 16 CF) and validation sets (17 AS, 20 CS; 16 AF, 23 CF) (Figure S1). Detailed demographic and clinical features of these enrolled volunteers are described in Table S1 and S2.
Beta-diversity rather than alpha-diversity exhibited a significant difference in PC
For salivary microbial diversity, the CP group exhibited a marked decrease when comparing the alpha-diversity with the PC group or the HC group as estimated by the following metrics: observed species, Chao1, and PD whole tree. No significant differences were observed between the PC group and the HC group in alpha-diversity (Figure 1(a)). A similar trend was also observed in fecal microbial diversity, but these indexes did not reach statistical significance (Figure 1(b)).
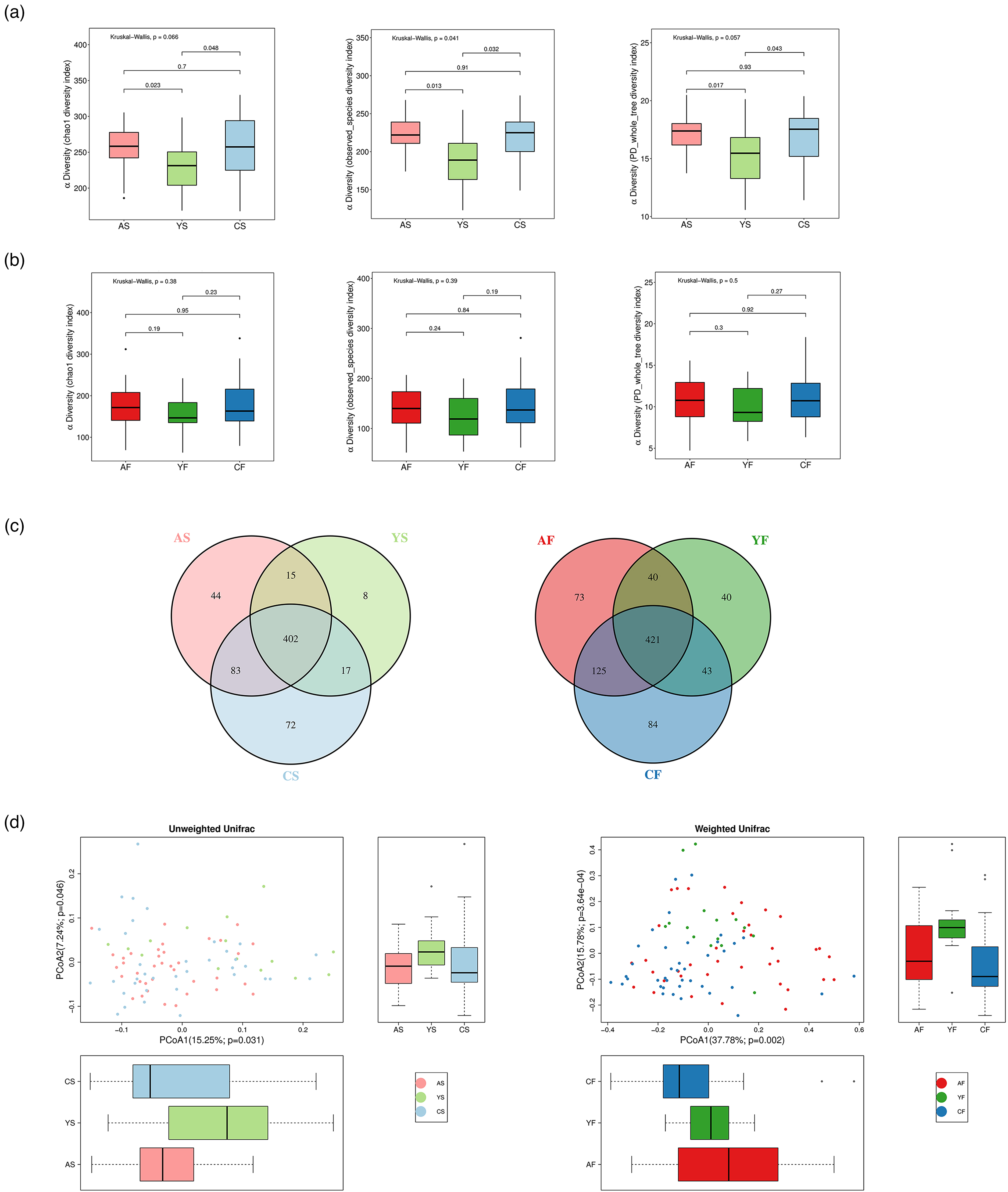
Beta-diversity rather than alpha-diversity showed a significant difference in PC. (a) For the saliva microbiome, α-diversity was lowest in the CP group, whereas no significance was observed between the PC group and the HC group. (b) A similar trend was also observed in fecal microbial diversity, but these indexes did not reach statistical significance. (c) Two Venn diagrams exhibiting the overlaps between groups revealed that 402 of 641 OTUs were shared among the three saliva groups, while 421 of 826 OTUs were shared among the three fecal groups. (d) β-diversity was computed with unweighted (left) or weighted (right) UniFrac by PCoA, showing that the microbial composition of the three groups was significantly different (P = 0.031, 0.046; P = 0.002, 3.64e−4).
In total, 1053 OTUs were obtained in 16S rRNA sequencing analysis, among which 826 OTUs were detected in fecal samples and 641 OTUs in saliva samples. An overlapping Venn diagram between the three groups showed that 40 OTUs were specific to PC and CP in feces and 15 OTUs in saliva. Notably, there were 73 fecal OTUs and 44 salivary OTUs specific to PC (Figure 1(c)). To present the microbial community space among samples, weighted and unweighted UniFrac analyses were adopted to calculate the beta-diversity, and PCoA indicated that the microbial composition was significantly different among the three groups (Figure 1(d)).
Differential analysis of the microbial composition among three groups
The (salivary or fecal) microbial compositions were discussed at the levels of phylum (Figure 2(a) and (b)) and genus (Figure 2(c) and (d)) among groups. Bacteroidetes, Firmicutes, and Proteobacteria were the most dominating phyla in all groups, together accounting for over 80% of the relative abundance on average.
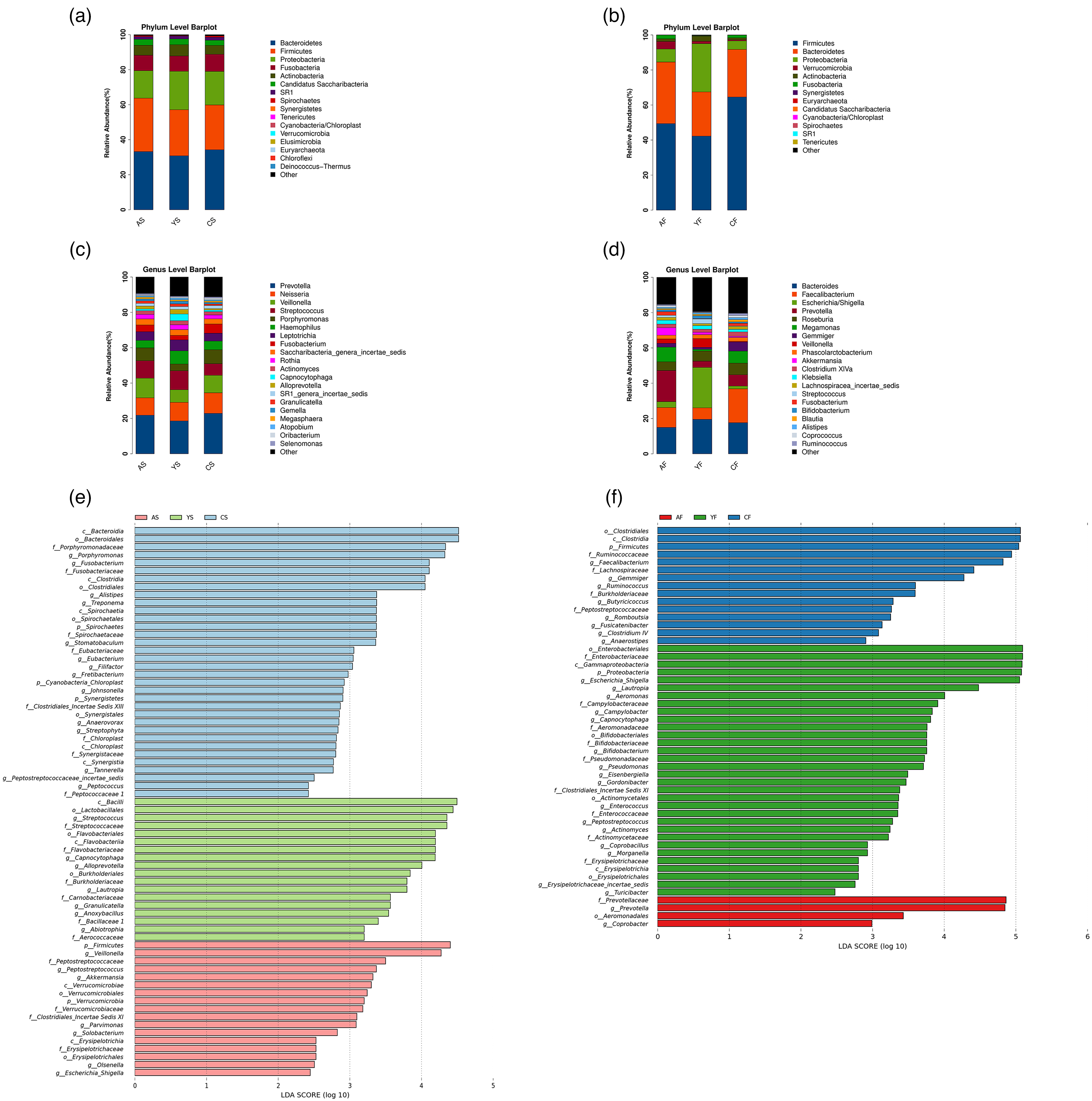
Differential analysis of salivary/fecal microbiota among the three groups. Relative abundance of saliva (left) and fecal (right) microbiota in the three groups at the levels of phylum ((a) and (b)) and genus ((c) and (d)). LEfSe analyses represent the differential microbial taxa of salivary (e) and fecal (f) microbiome among groups (P < 0.05, LDA score > 2). Modules enriched in PC are colored in red; modules enriched in CP are colored in green; and modules enriched in HC are colored in blue.
In terms of the oral microbiome, the average relative abundance of the two phyla Firmicutes and Verrucomicrobia in the PC group was significantly greater than that in the other two groups. Correspondingly, at the genus level, seven genera (Veillonella, Peptostreptococcus, Akkermansia, Parvimonas, Solobacterium, Olsenella, and Escherichia-Shigella) were significantly enriched (Figure 2(e)). Compared with that in the HC group, the average relative abundance of one phylum (Firmicutes) was significantly higher in both the PC and CP groups (Figure S2(a)). Specifically, four genera (Alloprevotella, Streptococcus, Granulicatella, and Solobacterium) were significantly enriched (Figure S2(b)). Notably, the differential oral genera were mostly symbiotic (Figure S4(a)).
In terms of the gut microbiome, compared with the other two groups, the average relative abundance of only two genera, Prevotella and Coprobacter, were significantly enriched in the PC group (Figure 2(f)). Compared with the HC group, only the Proteobacteria phylum abundance in the pancreatic disease group was significantly increased (Figure S3(a)). At the genus level, six genera (Peptostreptococcus, Actinomyces, Bifidobacterium, Campylobacter, Coprobacillus, and Escherichia-Shigella) were enriched in the pancreatic disease group (Figure S3(b)). The competition and synergy between different enterobacteria were not obvious (Figure S4(b)).
Exploring microbial markers of PC
To explore the potential of microbiomes as biomarkers, diagnostic models based on the oral or gut microbiome attempting to discriminate PC from HCs were then established. We found that the combination of 10 genera based on differential analysis of the oral microbiome could distinguish PC from HC with an AUC of 0.916 (95% CI 0.832-1) (Figure 3(a) and (b), and S5(a)), whereas a panel of 10 genera from fecal microbiome profiling could achieve an AUC of 0.856 (95% CI 0.74, 0.972) (Figure 3(c) and (d) and S5(b)).
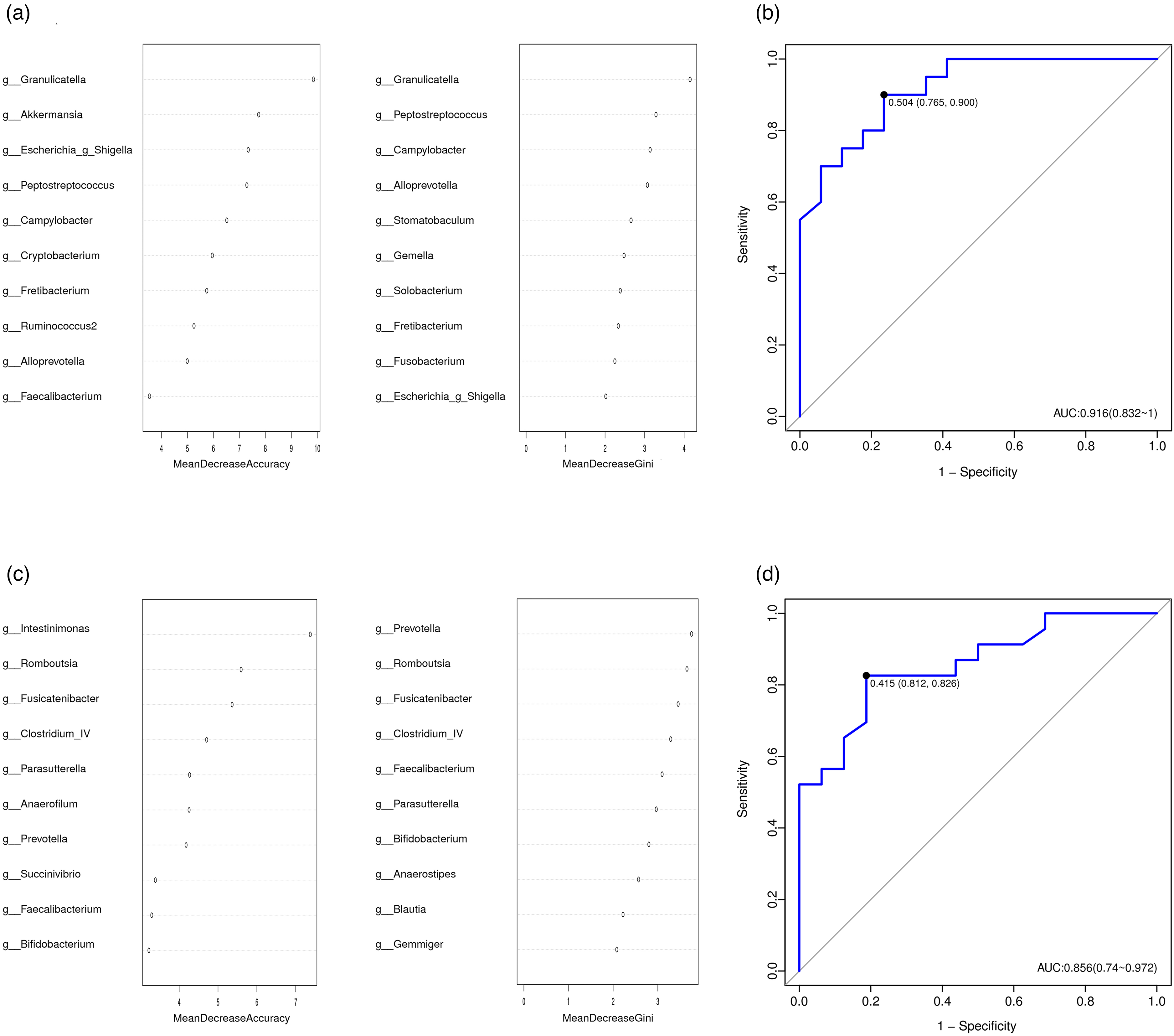
Identification of genus-based microbial markers of PC by RF algorithms. To obtain specific genera markers of PC, a 10-fold CV on two RF models (20 AS vs. 16 CS; 22 AF vs. 16 CF) was conducted in the discovery set. The importance of each genus in classification was ranked by mean decreasing accuracy and Gini coefficient for oral (a) and gut (c) microbiota. (b) A 10 oral genera panel could discriminate PC from controls with an AUC of 0.916 (95% CI 0.832-1). (d) A 10 gut genera panel could discriminate PC from controls with an AUC of 0.856 (95% CI 0.74, 0.972).
Obvious functional dysbiosis of microbiota in PC
To explore the functional changes in microbiota in the progression of PC, the PICRUSt method was adopted for functional analysis based on 16S rRNA data. The LEfSe method identified 5 oral microbiota-associated pathways (LDA score > 2, P < 0.05, Figure 4(a)) and 14 gut microbiota-associated pathways (LDA score > 2.5, P < 0.05, Figure 4(b)) involved in PC. Here, 16S function predicts that the genetic information processing pathways, such as mismatch repair, DNA replication, and homologous recombination, were increased in the oral microbiome of PC, whereas multiple amino acid metabolism pathways (alanine, aspartate, glutamate, and histidine metabolism) were weakened. In terms of gut microbiota, functional prediction additionally revealed obvious inflammatory pathway activation and decreased cell motility. Specifically, lipopolysaccharide biosynthesis and peptidoglycan biosynthesis were enhanced, whereas cytoskeleton biosynthesis was weakened.
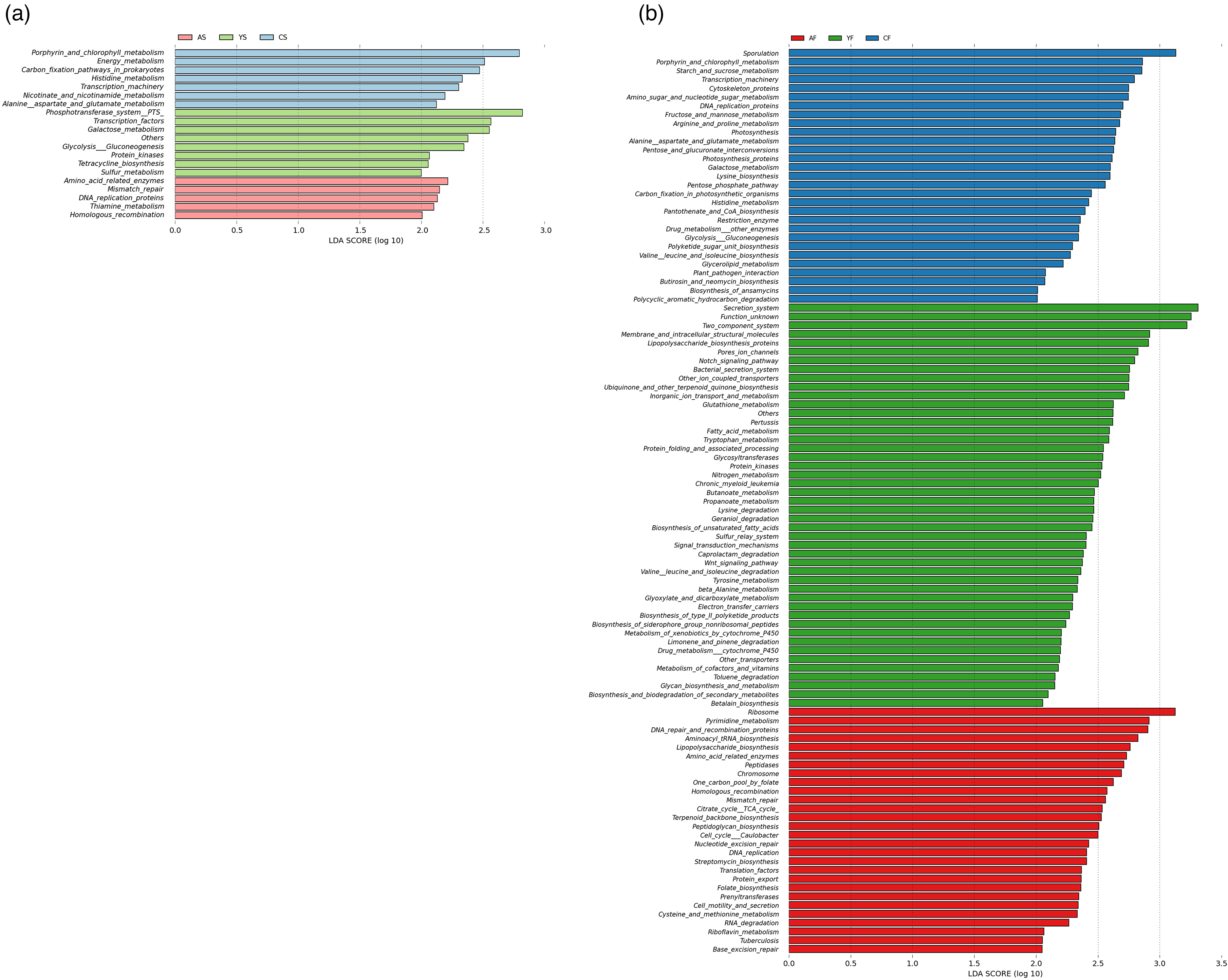
Functional prediction identified by LEfSe. (a) Five pathways based on the differential oral microbiota in the PC group were identified by the LEfSe method (P < 0.05, LDA > 2). (b) Fourteen pathways based on the differential gut microbiota in the PC group were identified by the LEfSe method (P < 0.05, LDA > 2.5). Modules enriched in PC are colored in red; modules enriched in CP are colored in green; and modules enriched in HC are colored in blue.
Discussion
Accumulating evidence has demonstrated that alterations in microbiota are associated with numerous diseases.19–21 In this article, we comprehensively described the oral and gut microbiota characteristics of a PC group in parallel via 16S rRNA analysis. Microbial diversity was lowest in the CP group, whereas no significance was observed between the PC group and the HC group. Notably, previous studies have reported a phenomenon similar to our findings: the diversity of microbiota is greater in the tumor group than that in precancerous group.12,22 Therefore, it is believed that an increase in the richness or diversity of the microbiota is not an indicator of health in our group but may hint at the overgrowth of diverse harmful or pathogenic bacteria in PC patients.
The microbiota composition changed significantly. Oral pathogenic genera (Granulicatella, Peptostreptococcus, Alloprevotella, Veillonella, Solobacterium, Streptococcus, etc.) exhibited a significant enrichment in patients with PC. In the gut, the abundances of bacterial genera such as Bifidobacterium and Butyricicoccus, known to include probiotics, were dramatically reduced, while opportunistic genera (Prevotella, Escherichia-Shigella, Peptostreptococcus, Actinomyces, etc.) were significantly enriched. Diagnostic models based on the characteristics of the oral or gut microbiota can effectively discriminate the PC samples in our study, indicating that microbial biomarkers have the potential to develop into noninvasive diagnostic tools for PC. Of the sample types, saliva could represent a superior sample type for PC microbial detection due to its advantages of convenient clinical collection, good patient compliance, and easy operation.
Based on the above analysis, we screened candidate genera that might be involved in the course of PC. Granulicatella contributed most to the effective discrimination of PC according to our Mean Decrease Accuracy and Mean Decrease Gini analysis (Figure 3(a)). Several Western studies have also found that the enrichment of Granulicatella adiacens in saliva is closely related to pancreatitis, intraductal papillary mucinous neoplasm (IPMN) and PC11,23,24; one of the studies specially revealed that the microbial community in pancreatic cyst fluid was symbiotic and enriched with oral bacteria, of which the burden of G. adiacens was particularly heavy and positively correlated with IPMN classification. The candidate gut genus was Prevotella, it was enriched in the PC group with an average relative abundance of 17.64%, which was significantly greater than that of the HC group (6.40%, P = 0.0009) and CP group (3.57%, P = 0.0147). Prevotella copri (P. copri) is typically the most common Prevotella in the gut. It has been found involved in the development of rheumatoid arthritis via the Th17/IL-17 pathway in mice.25–27 This pathway can also significantly accelerate the development of pancreatic intraepithelial neoplasia (PanIN, precancerous status), 28 so it is reasonable to suspect that P. copri also plays a role in driving PC through Th17 activation. In the future, it will be necessary to perform higher resolution sequencing analyses to identify PC-specific species and to further analyze their roles and mechanisms in the course of human PC.
PC microbiota were predicted to induce oncogenesis through a well-known LPS/TLR4 signaling pathway. 29 Besides, multiple metabolisms were also predicted to exhibit dysfunction in PC microbiome. For instance, arginine and proline metabolism were decreased, which could result in reduced level of polyamines, 30 and thus impair the maintenance of the intestinal barrier as well as the development of resident immune cells.31,32 The pyrimidine metabolism was enhanced in PC microbiome, which can be explained by the fact that increased pyrimidine biosynthesis is a corresponding result of cancer progression.30,33
Notably, several limitations of this study exist. First, 16S rRNA sequencing limits the interpretation of data in terms of resolution and functional analysis. Second, even though strict criteria have been established, diet, region, and other influencing factors cannot be completely eliminated in this study. To better address this issue, it is necessary to include an additional healthy control subgroup matched for smoking status/diabetes in the analysis of commensal microbiota. Third, although the number of patients with PC admitted to our hospital ranks first in China and these patients come from various regions, it is still necessary to perform additional multicenter studies to verify and summarize the findings of this paper. Fourth, although we did our best to minimize the environmental effect (e.g., sample collection, processing, and storage were performed according to the uniform protocols) participants were provided with sterile containers and instructed to retain specimens normatively, etc., we must acknowledge the recruitment of HCs from another hospital as a limitation that may have a potential effect on the results. Finally, it should be noted that we have confirmed the correlation between microbiota and PC in a PC group, but this study cannot provide direct evidence regarding whether a causal link exists between the two. Future research is necessary to investigate the role of PC-associated bacteria, especially Granulicatella and Prevotella, in the etiology of this disease.
Despite the limitations discussed above, the highlights of our study are as follows. First, it is a case-control study that provided a comprehensive description regarding altered commensal microbiota in a Chinese PC group. The description is convincing given that the commensal microbiota of Chinese patients with PC exhibits unique microbial signatures that are similar to Western studies. Second, this study further explored the possibility of exploiting microbial markers for the diagnosis of PC. Third, simultaneous collection of saliva and fecal samples from the same volunteers will favor the comparison of diagnostic efficacy between two sample types with minimum interference. Saliva appears to serve as a better clinical test sample than stool. Fourth, data analysis proposed the primary candidate genera and pathways that might be involved in the progression of PC, offering a new perspective on pathogenesis research to further promote early diagnosis along with timely treatment of PC.
Supplemental Material
sj-docx-1-jbm-10.1177_03936155231166721 - Supplemental material for Alterations of commensal microbiota are associated with pancreatic cancer
Supplemental material, sj-docx-1-jbm-10.1177_03936155231166721 for Alterations of commensal microbiota are associated with pancreatic cancer by Tian Chen, Xuejiao Li, Gaoming Li, Yun Liu, Xiaochun Huang, Wei Ma, Chao Qian, Jie Guo, Shuo Wang, Qin Qin and Shanrong Liu in The International Journal of Biological Markers
Supplemental Material
sj-xlsx-2-jbm-10.1177_03936155231166721 - Supplemental material for Alterations of commensal microbiota are associated with pancreatic cancer
Supplemental material, sj-xlsx-2-jbm-10.1177_03936155231166721 for Alterations of commensal microbiota are associated with pancreatic cancer by Tian Chen, Xuejiao Li, Gaoming Li, Yun Liu, Xiaochun Huang, Wei Ma, Chao Qian, Jie Guo, Shuo Wang, Qin Qin and Shanrong Liu in The International Journal of Biological Markers
Footnotes
Acknowledgments
We thank Prof. Gang Jin, Prof. Shiwei Guo, and Dr. Gang Nie at the Department of Hepatobiliary Pancreatic Surgery, Changhai Hospital, for their kind help during the sample collection.
Declaration of participant consent
Informed consent was obtained from all participants. They understand that their privacy rights will always be observed.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
T. Chen and X. Li contributed equally to this work.
Funding
The author(s) disclosed receipt of the following support for the research, authorship, and/or publication of this article: This study was partially supported by Shanghai Science and Technology Committe (Grant No.18XD1405300) and Air Force Hospital of Eastern Theater Command (Grant No.21YY003).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.