
Abstract
Background
The Gleason Score is well correlated with biological behavior and prognosis in prostate adenocarcinoma (PRAD). This study was derived to determine the clinical significance and function of Gleason-Score-related genes in PRAD.
Methods
RNA-sequencing profiles and clinical data were extracted from the The Cancer Genome Atlas PRAD database. The Gleason-Score-related genes were screened out by the Jonckheere-Terpstra rank-based test. The “limma” R package was performed for differentially expressed genes. Next, a Kaplan–Meier survival analysis was performed. Correlation MT1L expression levels with tumor stage, non-tumor tissue stage, radiation therapy, and residual tumor were analyzed. Further, MT1L expression was detected in PRAD cell lines by reverse transcription-quantitative polymerase chain reaction assay. Overexpression of MT1L was constructed and used for cell count kit-8, flow cytometric assay, transwell assay, and wound-healing assay.
Results
Survival analysis showed 15 Gleason-Score-related genes as prognostic biomarkers in PRAD. The high-frequency deletion of MT1L was verified in PRAD. Furthermore, MT1L expression was decreased in PRAD cell lines than RWPE-1 cells, and overexpression of MT1L repressed cell proliferation and migration, and induced apoptosis in PC-3 cells.
Conclusion
Gleason-Score-related MT1L may serve as a biomarker of poor prognostic biomarker in PRAD. In addition, MT1L plays a tumor suppressor in PRAD progression, which is beneficial for PRAD diagnosis and treatment research.
Introduction
Prostate cancer (PCa) is one of the most common malignant tumors in the world, accounting for about 14% of all tumors in males. 1 In western countries, the incidence rate of PCa is second and its mortality rate is sixth in males. 2 PCa includes prostate adenocarcinoma (PRAD), which is a pathological type of PCa and accounts for more than 98% of PCa. In recent years, the incidence rate of PRAD has increased with the aging population in China. Studies have shown that approximately 30% of the elderly over 65 years old are diagnosed with PRAD in the middle and late stages. 3 Although the prognosis of most PRAD patients is relatively good, the recurrence or metastasis after treatment is poor. 4 At present, serum prostate specific antigen (PSA) level, Gleason Score, and tumor stage are the most important prognostic factors of PRAD, and play an important role in guiding clinical diagnosis and treatment decisions. However, these prognostic factors are still inaccurate in predicting the progression of PRAD.
The Gleason scoring system was developed by Gleason in 1966 based on 2911 PCa specimens and represents the specific characteristics of PCa tissue growth. 5 Combined with the disease progression and survival data after radical prostatectomy, the tissues were classified into 5 grades: 1 = good, and 5 = poor. The Gleason Score is most widely used as a pathological grading method for PCa and is closely related to the biological behaviors of PCa. 6 The Gleason Score has certain advantages over indicators in predicting tumor stage, and can predict the treatment effect on the tumor. Currently, the Gleason Score is an important reference for the clinical treatment of PCa 7 ; however, it cannot accurately judge the prognosis of PCa and predict its biological behavior. 8 Therefore, there is still a lack of effective predictors to guide clinical practice, diagnosis, and treatment decision-making.
In recent years, the molecular typing of cancer has been studied in many kinds of tumors, such as breast cancer, leukemia, and gastrointestinal cancer.9,10 Tumor classification has changed from traditional morphology to “molecular typing” based on molecular characteristics. The research of tumor molecular typing plays an important role in guiding clinical decision-making and prognosis. 11 The key step of tumor molecular typing is to find more effective molecular typing and to identify markers related to tumor prognosis. 12 Therefore, it is urgent to find prognostic molecular markers related to the Gleason Score to evaluate the prognosis and treatment of PCa.
MT1L belongs to the family of metallothioneins (MTs). 13 MTs are small cysteine-rich proteins, which, in humans, are encoded by multigene family on chromosome. 14 MTs are involved in diverse processes, including metal homeostasis and detoxification, the oxidative stress response, and cell proliferation. 15 Numerous gene expression analyses have revealed that the change of MT expression is correlated with the developmental progression of various malignancies.16,17 Previous studies have found that MT1L regulates the immune microenvironment and is related to poor prognosis in bladder cancer. 18 The MT1L gene serves as a promising outcome predictor and a potential therapeutic target in colorectal cancer. 19 The clinical significance and role of MT1L in PCa is still unclear.
In this study, the Gleason-Score-related genes were identified by the Jonckheere-Terpstra Test based on RNA-sequencing and the clinical data of The Cancer Genome Atlas (TCGA) PRAD. Further, we analyzed the survival and clinical significance of Gleason-Score-related genes in PRAD. PCa cell lines (DU-145 and PC-3) were cultured and transfected with Gleason-Score-related genes (MT1L) to explore the function of MT1L in vitro. The aim of this study was to determine the clinical significance and function of Gleason-Score-related genes (MT1L) in PRAD.
Materials and method
Data acquisition
RNA-sequencing profiles and clinical data were extracted from the TCGA PRAD database (https://cancergenome.nih.gov/). The Gleason-Score-related genes were screened out by the Jonckheere-Terpstra-Test-based clinical information and RNA-sequencing. The “limma” (linear models for microarray data) R package was performed for differentially expressed genes (DEGs) identifying between tumor and normal samples. 20 DEGs were identified by the standard of the absolute value of Log2|fold change (FC)| ≥ 1, and P value ≤ 0.05.
Gene expression profiling interactive analysis
The online database GEPIA (http://gepia.cancer-pku.cn) provided interactively analyses of cancer and normal genes from TCGA. 21 The expression of 15 Gleason-Score-related genes (ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1) were analyzed in tumor (T) group and adjacent non-tumor tissue (N) group of the PRAD TCGA dataset. To further clarify the relationship between 15 Gleason-Score-related genes (ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1) expression and PCa prognosis, GEPIA was used to explore survival curves, including disease-free survival (DFS), which were based on log rank for statistical analysis.
Copy number alteration assay
The cBioPortal Web resource (http://www.cbioportal.org/) contains the data of 126 tumor genome research studies, including large-scale tumor research projects such as TCGA and ICGC. The copy number alteration (CNA) of 15 Gleason-Score-related genes (ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1) was analyzed by the cBioPortal database.
Cell culture
Human normal prostate epithelial cells (RWPE-1) and the PCa cell lines (DU-145, and PC-3) were purchased from ATCC (Manassas, VA, USA). RWPE-1 cells were cultured in K-SFM medium containing 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and added 0.05 mg/mL BPE, 5 ng/mL EGF. DU-145 and PC-3 cells were cultured in RPMI 1640 medium (RPMI-1640; Gibco) containing 10% FBS in an incubator with 37°C and 5% CO2 saturation humidity.
Cell transfection
The overexpression plasmid of pcDNA3.1-MT1L and control vector pcDNA3.1 were purchased from GenePharma (Shanghai, China). For cell transfection, PC-3 cell were transfected with pcDNA3.1-MT1L or empty vector using LipofectaminTM2000 (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's instructions.
Reverse transcription-quantitative polymerase chain reaction
Total RNA was extracted from cells after relevant treatment with TRIzol reagent (Invitrogen, USA). The cDNA was synthesized by using The Reverse Transcription Kit was used to synthesize the cDNA according to the manufacturer's instructions. The cDNA was amplified by RT-qPCR using AceQ Universal SYBR qPCR Master Mix on an ABI 7500 PCR system. Data analyses for gene expression were performed using the 2−ΔΔCt method. GAPDH was used as an internal control. The primer sequences following: GAPDH-F: 5′-GAAGGTGAAGGTCGGAGTC-3′, GAPDH-R: 5′-GAAGATGGTGATGGGATTT-3′; MT1L-F: 5′-AGTGCAAATGAACCTCCTGC-3′, MT1L-R: 5′-ATCCAGGTTTGTGCAGGTTG-3′.
Cell proliferation assay
Cell proliferation was analyzed by cell count kit 8 (CCK-8, Dojindo, Japan) assay after relevant treatment. In brief, logarithmic growth cells (1×105) were seeded into 96-well cell culture plates with 4 repented wells for each treatment. At 0, 24, 48, and 72 h, 10μL CCK-8 solution was added into each well the manufacturer's instructions and were incubated for 1h. The absorbance of each well was determined by enzyme-labeled instruments (Molecular Devices, San Jose, CA, USA) Microplate Auto reader at 450 nm. The experiment was performed three times in triplicate.
Flow cytometry assay
The cell apoptosis was detected using the annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer's instructions. In brief, PC-3 cells were washed with phosphate buffer saline (PBS) after relevant treatment and transfection. Then, 500 μL binding buffer were resuspended cells, 5μl Annexin V-FITC and 5 μL PI were stained cells at room temperature in the dark for 15 min. Finally, the cell apoptotic rate was analyzed by a FACScan® flow cytometer (BD Biosciences) determined using software FlowJo (7.6.1; FlowJo LLC).
Transwell assay
The PC-3 cells were digested and re-suspended in serum-free 1640 medium at 48 h after transfection. Cells were counted and 3×104 were inoculated into 24-well plate transwell chamber. In the lower chamber the medium was added with 10% FBS. After 24 h of culture, the chamber was taken out and fixed with 10% neutral formalin. The cells that did not pass through the chamber membrane were wiped with a cotton swab. The migration cells were stained with crystal violet for 20 min. The migration cells were photographed under the microscope from four randomly selected visual fields.
Wound-healing assay
The cell migration was analyzed by a wound-healing assay. In brief, The PC-3 cells were seeded in 6-well cell culture plates for each treatment. When the cells had grown to 100% in confluency, the tip of the 200 μL pipette was used to make a scratch on the monolayer. PBS was applied to wash off the scribed cells three times, and a serum-free medium was added. Cell migration was monitored under a microscope and then photographed at 0 and 48 h. The experiment was performed three times in triplicate.
Statistical analysis
The data collected from three independent experiments were expressed as the mean ± SD. The GraphPad Prism 8.0 was used to perform the statistical analyses. Paired Student's t-test or one-way analysis of variance (ANOVA) were used for analysis with multiple comparisons. P<0.05 was considered statistically significant.
Results
Gleason-Score-related genes were identified in prostate adenocarcinoma
To explore the biomarkers related to the Gleason Score in PRAD, we downloaded the mRNA expression data and clinical information from TCGA database. We screened out 100 genes that correlate with the Gleason Score in PCa by R software. A volcano plot showed that the identification of 100 genes was significantly positively/negatively related to the Gleason Score (Figure 1(a)). A heat map showed that 50 genes were positively related to the Gleason Score in PRAD, including TROAP, UBE2C, and more (Figure 1(b)). The heat map in Figure 1(c) shows that 50 genes were negatively related to the Gleason Score in PRAD, including SLC7A4, AC020571.3, RPE65, and more. Further, 32 genes with a correlation coefficient more than 0.4 or less than −0.4 were selected to verify the expression difference in a PRAD tumor samples compared with normal samples.
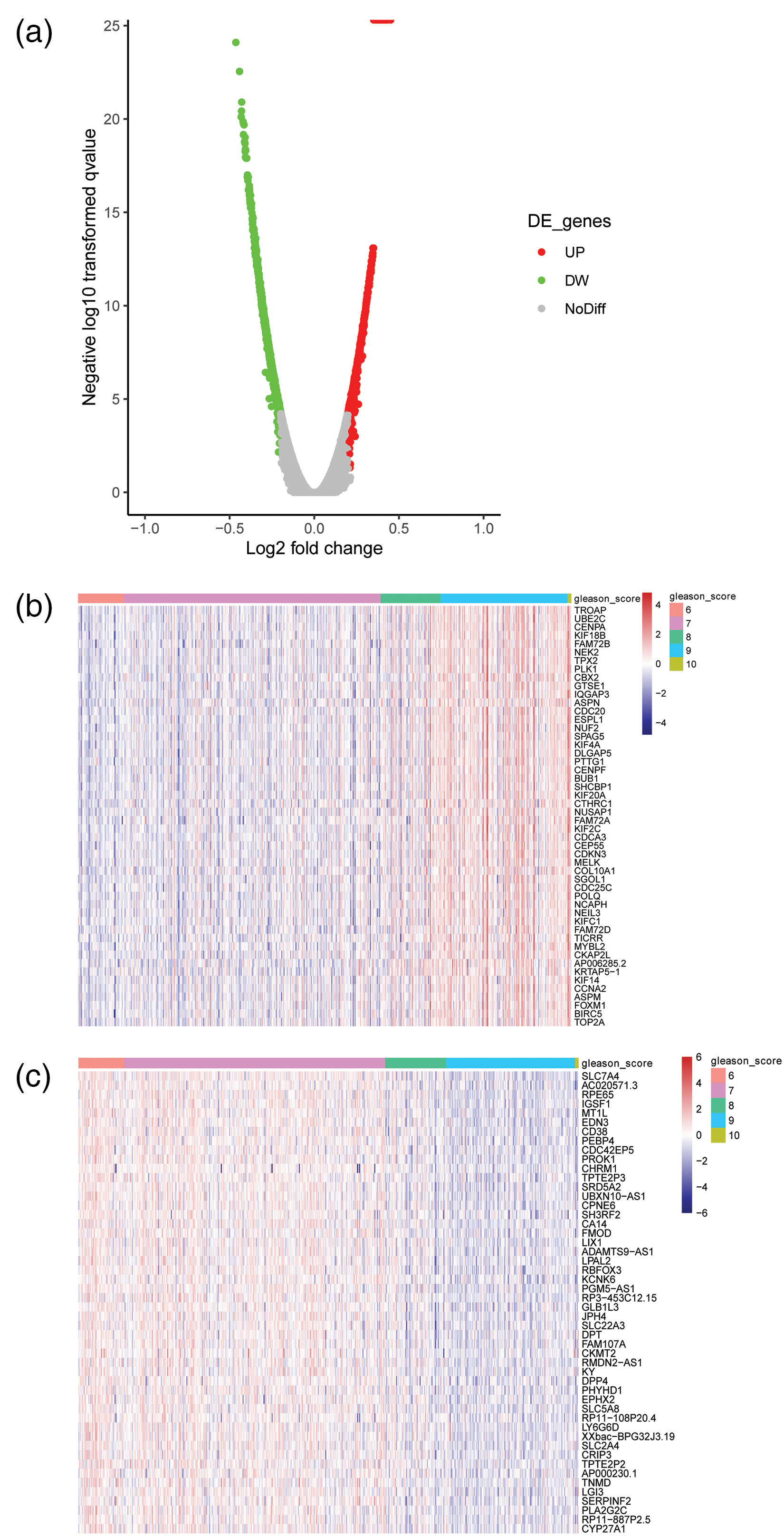
Identification of Gleason-score-related genes in TCGA PRAD. (a) A volcano plot for Gleason-Score-related genes in PRAD. Red dots represent significantly positive related genes; green dots represent significantly negatively correlated genes; and grey dots indicate not significant. (b) A heatmap of 50 genes positively related to the Gleason Score in PRAD. (c) A heatmap of 50 genes negatively related to the Gleason Score in PRAD.
The expression of 15 Gleason-Score-related genes were analyzed in PRAD
Next, 32 Gleason-Score-related genes were identified based on the standard of correlation coefficient >0.4 or <0.4 in the PRAD TCGA dataset. To analyze the DEGs in PRAD, we downloaded the mRNA expression data from TCGA database. The 7070 DEGs were identified in PCa by using the R package DEseq2 based on change fold ≥2 and a P-value <0.05. Further, a Venn diagram showed that 15 common genes were enriched between 32 Gleason-Score-related genes and 7070 DEGs in PRAD (Figure 2(a)), including ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1. The cluster analysis suggested that there were significant differences in PRAD tumor samples and normal samples (Figure 2(b)).
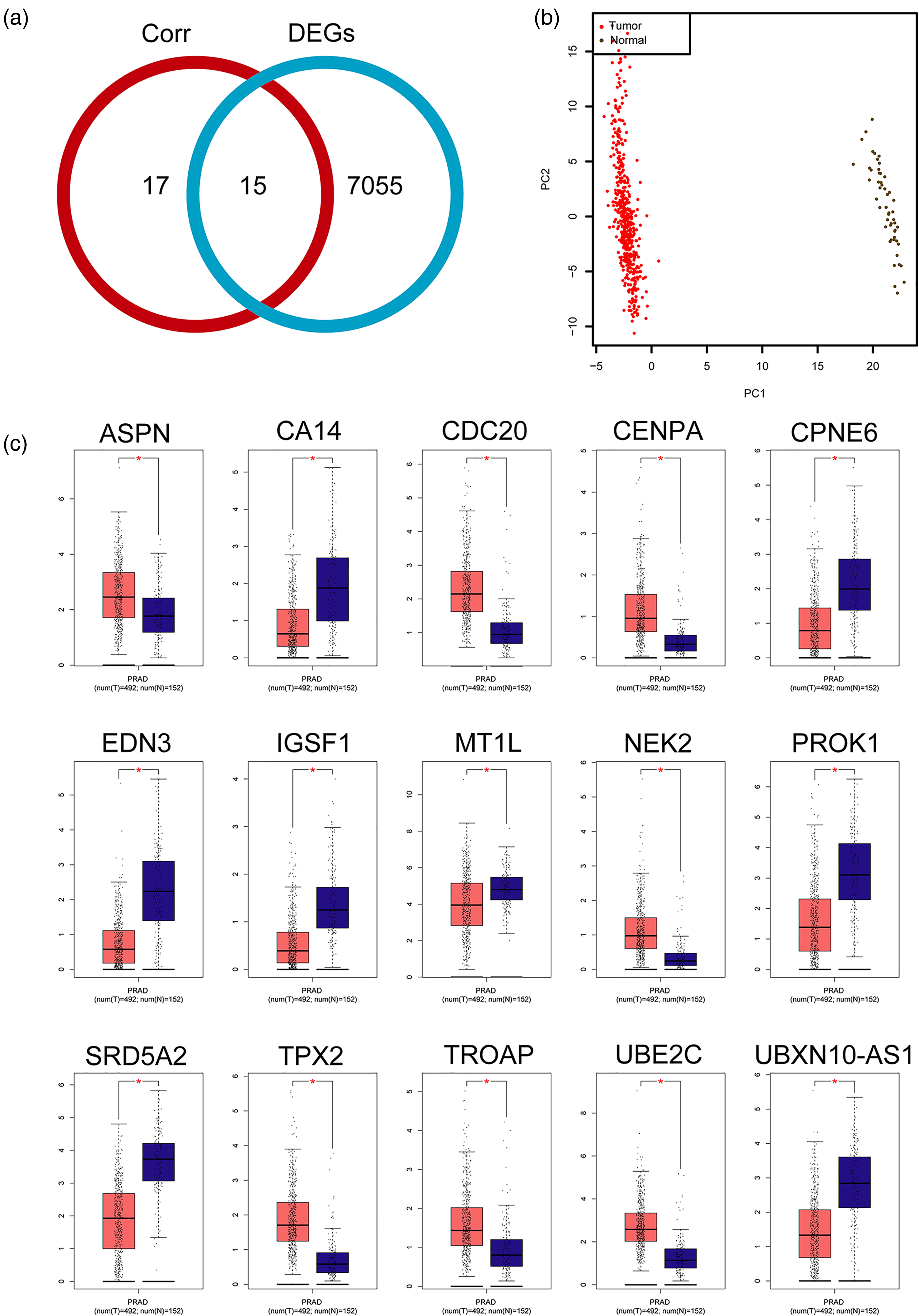
The expression level of 15 genes related to the Gleason Score in PRAD. (a) A Venn diagram showing that 15 common genes in the Corr group (Gleason-Score-correlated genes) and DEGs groups. Red circle indicates 32 genes of correlation coefficient >0.4 or <0.4. Blue circle indicates 7070 DEGs. Corr 17 genes and 7055 DEGs indicate non-common genes between the correlation group and the DEGs group. (b) Cluster partition of PRAD tumor and PRAD normal. (c) Box plot showing that the expression level of ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1 in the PRAD tumor group and the PRAD normal group from the GEPIA database.
Next, we verified the expression of 15 Gleason-Score-related genes in PRAD tumor samples compared to normal samples from the GEPIA database. As shown in Figure 2(c), the expression levels of ASPN, CDC20, CENPA, NEK2, TPX2, TROAP, and UBE2C were significantly increased in PRAD tumor samples compared with normal samples. The expression levels of CA14, CPNE6, EDN3, IGSF1, MT1L, PROK1, SRD5A2, and UBXN10-AS1 were significantly decreased in PRAD tumor samples compared with normal samples.
The DFS analysis of 15 Gleason-Score-related genes in PRAD
To explore the prognosis value of 15 genes in PRAD, we analyzed the DFS in PRAD. Survival analysis showed that the high expression group of ASPN, CDC20, CENPA, NEK2, TPX2, TROAP, and UBE2C were significantly associated with a shorter time of DFS than the low expression group in PRAD. The low expression group of CA14, CPNE6, EDN3, IGSF1, MT1L, PROK1, SRD5A2, and UBXN10-AS1 were significantly associated with shorter time of DFS than the high expression group in PRAD (Figure 3). These results demonstrate that the 15 Gleason-Score-related genes are prognostic biomarkers in PRAD.
Correlation between clinical–pathological parameters and MT1L expression in PRAD
Next, the CNA of ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1 was analyzed by the cBioPortal database. As shown in Figure 4(a), the CNA frequency of ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, TPX2, NEK2, TROAP, PROK1, IGSF1, UBE2C, PLA2G2C, MT1L, and SRD5A2 was 0.6%, 1.8%, 0.6%, 0.8%, 0.8%, 1%, 0.8%, 1%, 0.8%, 0.8%, 1.4%, 0.8%, 1%, 2.8%, and 1.2%, respectively. Interestingly, the CNA frequency of MT1L was higher than other Gleason-Score-related genes in PRAD; the alteration frequency of MT1L was 2.8%. The clinical value of the MT1L expression level in PRAD had not been characterized. Therefore, we further analyzed the clinical significance and biological function of MT1L expression in PRAD.
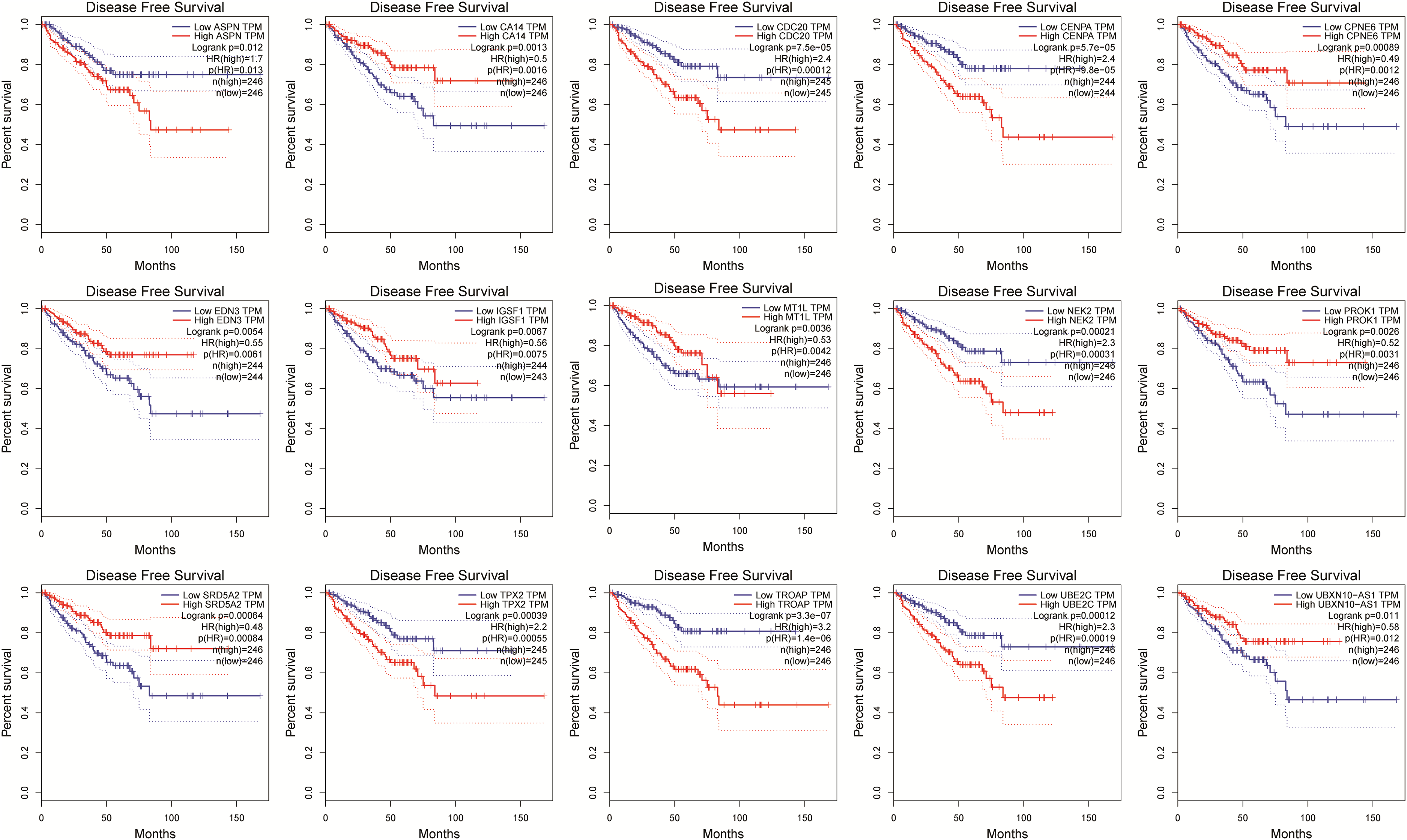
Survival analysis of 15 genes related to the Gleason Score in PRAD.
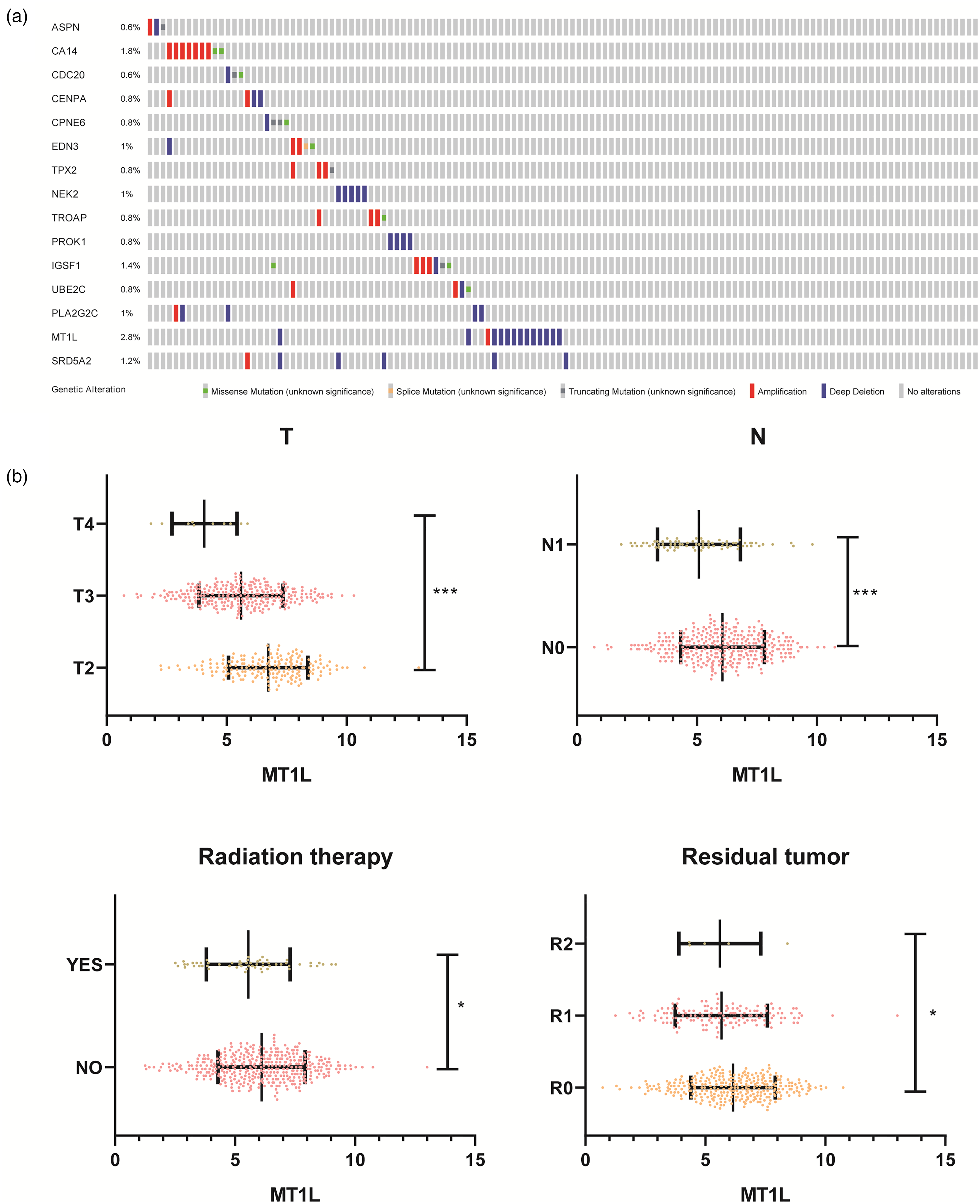
Correlation between clinical–pathological parameters and MT1L expression in PRAD. (a) The copy number alteration of 15 genes related to the Gleason Score were analyzed. (b) Association between MT1L expression level and T stage, N stage, radiation therapy, and residual tumor in PRAD.
We explored the MT1L expression level and its correlation between clinical–pathological parameters in PRAD by using GEPIA and R software based on the data downloaded from TCGA database. The MT1L expression level significantly negatively correlated with T stage, N stage, radiation therapy and residual tumor (Figure 4(b)). Further, the MT1L expression levels were significantly decreased in T4 vs. T3; T3 vs. T2; N1 vs. N0; the therapy group vs. no therapy group; R2 vs. R1; and R1 vs. R0 (Figure 4(b)). The results showed that MT1L has excellent diagnostic value in PRAD patients.
Overexpression of MT1L inhibited cell proliferation and migration and induced apoptosis in PC-3 cells
The biological function of MT1L in PRAD is unclear, so we investigated the expression and function of MT1L in RWPE and PCa cell lines (DU-145, and PC-3). First, the RT-qPCR assay showed that the expression of MT1L was significantly reduced in DU-145 and PC-3 cells compared with RWPE-1 cells (Figure 5(a)). The overexpression plasmid of pcDNA3.1-MT1L and control vector pcDNA3.1 were transfected into PC-3 cells, and RT-qPCR verified that the overexpression of MT1L in the OE-MT1L group compared with the control group (Figure 5(b)). A CCK-8 assay revealed that overexpression of MT1L significantly inhibited the cell proliferation in PC-3 cells (Figure 5(c)). Second, we analyzed the effect of MT1L on the apoptosis of PC-3 cells by using flow cytometry. We found that the overexpression of MT1L induced the cell apoptotic rates compared with the control (Figure 5(d)). Further, a transwell assay showed that the up-regulation of MT1L markedly decreased the invasion of PC-3 (Figure 5(e)). Also, the wound-healing assay revealed that up-regulation of MT1L significantly decreased the cell migration in PC-3 cells for 72 h (Figure 5(f)). Therefore, these results revealed that overexpression of MT1L markedly inhibited cell proliferation and migration, and promoted cell apoptosis in PC-3 cells.
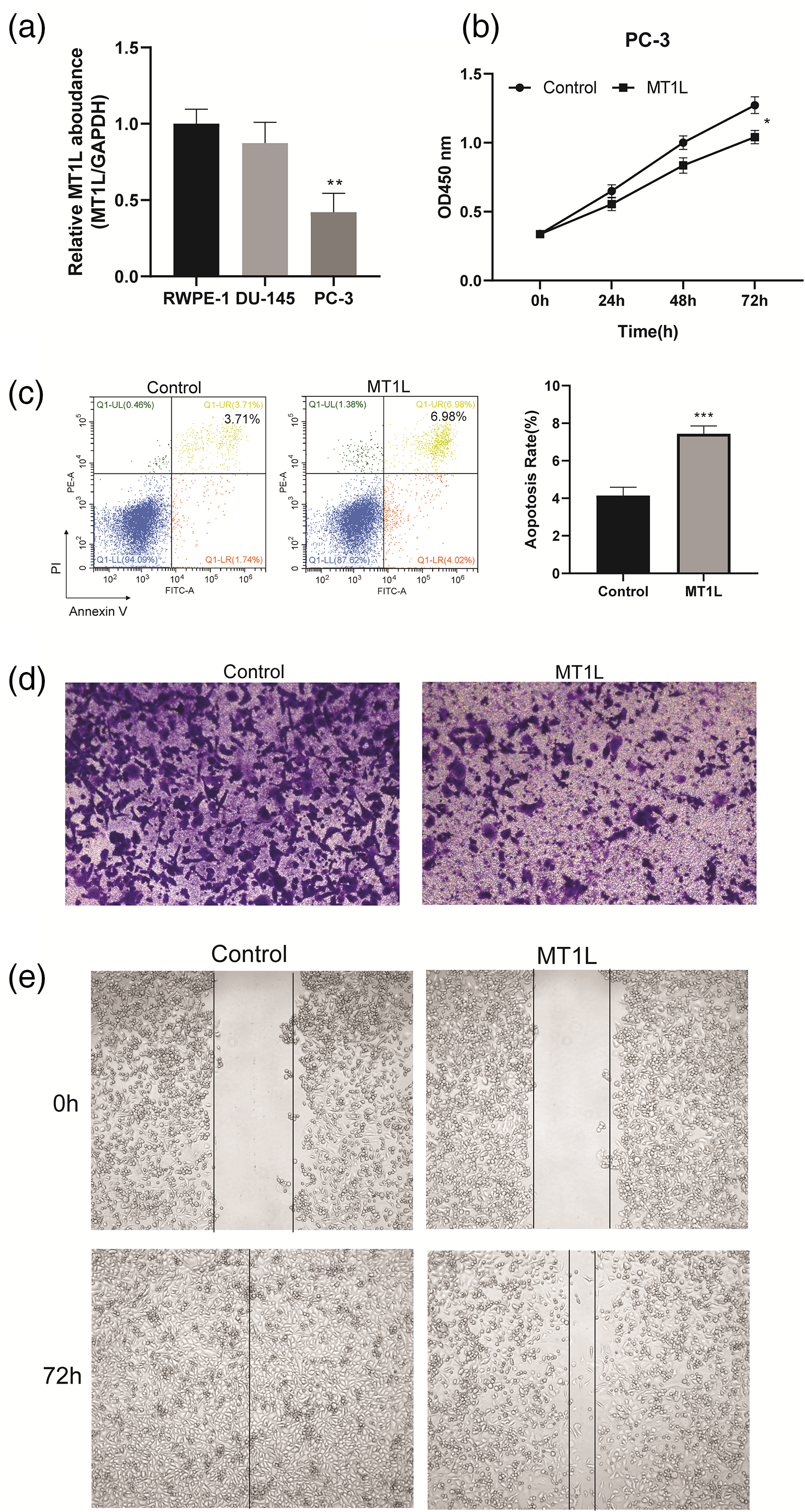
The effect of overexpressed MT1L on proliferation, apoptosis, and migration of PC-3 cells. (a) RT-qPCR was used to detect the expression level of MT1L in RWPE-1, DU-145, and PC-3. (b) CCK-8 assay was used to analyze the effect of overexpressed MT1L on PC-3 cell proliferation. (c) Flow cytometry was used to analyze the effect of overexpressed MT1L on PC-3 cell apoptosis. (d) Transwell migration assay. (e) Wound-healing assay was used to detect the effect overexpressed MT1L on PC-3 cell apoptosis. Data are represented as mean ± SEM. (*P<0.05, **P<0.01, ***P<0.001).
Discussion
PCa is one of the most common malignant tumors in the male genitourinary system. 22 The Gleason grading system is the best grading system that reflects the correlation between the pathological characteristics of PCa and its biological behavior.23,24 It is an important indicator for formulating treatment plans and evaluating prognoses for patients with PCa.22,25 PRAD is 98% of PCa. In this study, we identified that 100 genes were significantly positively/negatively related with the Gleason Score in PRAD, including ASPN, CA14, CDC20, and MT1L, and more. Further, we showed that 15 common genes were enriched between the Gleason-Score-related genes and DEGs in PRAD, including ASPN, CA14, CDC20, CENPA, CPNE6, EDN3, IGSF1, MT1L, NEK2, PROK1, SRD5A2, TPX2, TROAP, UBE2C, and UBXN10-AS1.
The Gleason Score is the best predictor of PCa prognosis in patients with localized disease. 26 It is one of the most important indexes to select and judge the prognosis of PCa. 27 Previous studies suggested that five gene signatures related to the Gleason Score serve as novel biomarkers for identifying early recurring events and contribute to the early diagnosis of PRAD. 28 The relative gene expression of CDH1, ESR2, VDR, and SRD5A2 correlated with the differentiation status of tumor Gleason Scores in PRAD. 29 In this study, the survival analysis showed that the high expression group of ASPN, CDC20, CENPA, NEK2, TPX2, TROAP, and UBE2C were significantly associated with a shorter time of DFS than the low expression group in PRAD. The low expression group of CA14, CPNE6, EDN3, IGSF1, MT1L, PROK1, SRD5A2, and UBXN10-AS1 were significantly associated with a shorter time of DFS than the high expression group in PRAD. Previous studies have found that IGSF1 and CA14 are possible new early markers in PCa.30,31 The Zhang et al. 32 study found that Asporin (ASPN) and EDN3 are possibly novel diagnostic indicators of PCa, whereas the high expression of ASPN is associated with worse prognosis of PCa. CDC20 is important biomarkers and potential therapeutic targets for metastatic PCa. 33 Previous studies have reported that CENPA, NEK2, TPX2, and UBE2C had high expression in PCa and contribute to its progression.34–38 The SRD5A2 genetic variation was associated with the risk of treatment failure in abiraterone and augmented the ability of prognostic stratification in PCa. 39 Moreover, our results have demonstrated that the 15 Gleason-Score-related genes are prognostic biomarkers in PRAD.
MT1L is a member of the MTs family.13,40 Studies have shown that MT1L plays a tumor regulatory role in the process of tumorigenesis. It can regulate cell growth and proliferation, protect the body from the negative effects of oxidative stress, antitumor drugs, and radiation by combining heavy metals such as zinc and copper.40,41 Recent studies suggest that the pseudogene MT1L regulates the immune microenvironment, correlates with poor survival, and is an independent prognostic biomarker in bladder cancer. 18 In this study, we found that the MT1L expression level significantly negatively correlated with T stage, N stage, radiation therapy, and residual tumor in PRAD. Overexpression of MT1L markedly inhibited cell proliferation and migration, and promoted cell apoptosis in PC-3 cells.
Conclusion
Overall, we screened out the Gleason-Score-related genes in PRAD based on TCGA database. Then the high frequency deletion of MT1L was identified. MT1L has excellent diagnostic value in PRAD patients. The cellular experiments suggested that MT1L is a tumor suppressor while overexpression of MT1L markedly inhibited cell proliferation and migration, and promoted cell apoptosis in vitro. This study provides a better understanding for a potential biomarker and an underlying mechanism in PRAD, which is beneficial for PRAD diagnosis and treatment research.
Footnotes
Acknowledgement
We thank the authors of TCGA for making their data public for analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Beijing health promotion association urology research foundation.
Data availability statement
The analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.