
Abstract
Raeder's syndrome was first described by the Norwegian ophthalmologist J. G. Raeder in 1918, and the description extended in 1924 by the same author. The seminal report was a description of a young, male patient with unilateral periocular pain combined with ipsilateral miosis and ptosis, and with slight objective signs of trigeminal nerve involvement. Autopsy demonstrated a tumor at the base of the skull in the middle cranial fossa. The term “paratrigeminal” was coined for the picture reported. Later case reports by the same and other authors have included patients with a more benign clinical course, including spontaneous remissions, with unilateral periocular pain and ipsilateral signs of oculosympathetic paresis as the common denominator. This review is a chronological survey of the main contributions that have appeared in the literature and an outline of the various definitions of the syndrome, including a recent classification as well as some pathophysiological and prognostic considerations.
When we see patients with unilateral periocular pain in our clinical practice, Ræder's syndrome will be one diagnostic possibility. The syndrome was first described in 1918 by the Norwegian ophthalmologist Georg Ræder (1), and later extended in 1924 by the same author (2). Since then, several reports have appeared, often describing cases with a clinical picture at some variance with Ræder's original cases. Consequently, there has been some controversy about the definition of the condition, and hence about its clinical significance and how patients should have their diagnostic work-up. In this short presentation, I will briefly review some main contributions that have been made, applying some kind of chronological order and ending up with a proposal of the current view.
The very original paper was published in 1918 in the Norwegian Magazine of Medical Science, and was a clinical description of a male 18-year-old patient who presented with pain in and around the left eye, the pain extending into the left temple and in fact also to the occiput (1). In addition, he suffered from anorexia, frequent vomiting and loss of weight. By examination, a left-sided miosis (difference 2.0 mm) and ptosis was disclosed, both pupils reacting normally to light stimulation. The lower jaw deviated to the left on opening the mouth, indicating a paresis of the left pterygoid muscles and accordingly involvement of the 3rd division of the left trigeminal nerve. As a matter of fact, Ræder also described a right deviation of the palate during intonation, suggesting involvement of the left glossopharyngeal nerve.
During the hospital stay, the patient stated that the pain also radiated to the left cheek and to “all teeth of the left side”, and mentioned that he sometimes had diplopia. There was tearing from the left eye. Cocaine eye drops failed to dilate the left pupil, strongly suggesting an oculosympathetic paresis. However, sweating in the face was intact, contrary to what would be expected in a full-blown Horner's syndrome. Ræder's conclusion was that there was an oculosympathetic paresis due to involvement of the sympathetic fibers distal to the bifurcation of the common carotid artery, since sweat fibers were thought to accompany the external carotid artery while the fibers destined for the pupil and eyelids probably were part of the plexus around the internal carotid artery. The involvement of the trigeminal nerve and of oculomotor nerves (diplopia) suggested a lesion in the middle cranial fossa adjacent to the ganglion Gasseri.
A few months later, the condition of the patient worsened, and he died, presumably due to pneumonia. The autopsy demonstrated a tumor at the base of the skull, between the ganglion Gasseri laterally and the hypophyseal stalk medially, the forward extension reaching the superior orbital fissure (Fig. 1). The oculomotor and the abducens nerves were deviated by the tumor, riding over it, thus accounting for the diplopia. The internal carotid artery was encroached by the tumor, accounting for the involvement of the sympathetic fibers. The Gasserian ganglion was pushed laterally, explaining the weakness of pterygoid muscles. The histological description concluded with a probable endothelioma.

Autopsy finding of tumor in the left middle cranial fossa in Ræder's original case. Drawing made by Ræder (1). The hatched area indicates the localization of the neoplasm at the base of the skull. Cranial nerves are indicated in roman numerals.
The sympathetic ganglia in the neck were not involved, and large tuberculous cavernes in both lungs did not seem to affect the brachial nerve plexus nor the sympathetic paravertebral chain. Accordingly, a preganglionic cause of Horner's syndrome seemed to be ruled out.
Ræder proposed a paratrigeminal lesion as a cause of the partial Horner's syndrome (i.e. the pure oculosympathetic paresis), the associated pain and the involvement of the trigeminal ganglion or nerve and oculomotor nerves.
In 1924, Ræder published this case in Brain along with four other cases (2). Four of the five cases had diminished sensation in the trigeminal area of the pain side, two had involvement of oculomotor or abducens nerves. All had miosis and all but one had ptosis, sweating in the face was intact when examined. Two cases were secondary to head trauma, one probably had trigeminal neuralgia, and no cause was found in one. Ræder gave little or no information as to the further clinical course and outcome.
Accordingly, Ræder's cases demonstrate a clinical picture of unilateral periocular pain associated with ptosis and miosis and some sign of involvement of the trigeminal nerve, sometimes also with affection of the oculomotor or abducens nerves. Ræder claimed that this combination of symptoms and signs indicated a paratrigeminal location and that it should prompt a thorough search for serious pathology in the middle cranial fossa. Nevertheless, four of his five cases seem to have had a favorable course, at least with no fatal outcome reported.
Many years were to pass until new cases were reported in the English literature. Bedrossian's case published in 1952 had left-sided pain in the eye and forehead with miosis, ptosis and diminished corneal sensation (3). After a dental abscess of the left upper lateral incisor was removed, all symptoms and signs disappeared within a week.
The Klingon and Smith's case reported in 1956 was disparate in that this was a woman who had anhidrosis in the ipsilateral part of the forehead (4). She also had a purulent nasal discharge and experienced a complete remission of pain within a few weeks. The authors object to the term “paratrigeminal”; no wonder, since the findings in their case were quite different from the symptoms and signs of Ræder's cases.
Smith added a new dimension to the syndrome, eventually leading to dilution and consequent confusion, ending up in later attempts to subdivide the syndrome. In 1958, he reported eight male cases with unilateral headaches and a partial Horner's syndrome, none of them demonstrating involvement of other cranial nerves (5). In spite of this lack of involvement of the trigeminal or other parasellar cranial nerves, he coined the term Ræder's syndrome for his cases, thus in many ways redefining the syndrome. The clinical course was favorable with disappearance of the pain usually within a few months. Retrospectively, some of his patients presumably were cases of cluster headache, which Smith himself admits by saying “it is possible that Dr. Horton's cases are similar to those mentioned in this report and that the only difference is in terminology”.
Boniuk and Schlezinger extended this redefinition, proposing a formal division of Ræder's syndrome into Group I and Group II cases (6). Group I is the Ræder's syndrome proper, with involvement of cranial nerves in addition to the pain and the oculosympathetic paresis, while Group II is merely a painful postganglionic Horner's syndrome. Their 9 patients all were Group II cases, with normal sensation of the face and no oculomotor involvement and with normal facial sweating. They were all males, and complete recovery within weeks or a few months was the rule as far as pain was concerned. The conclusion was that Group II Ræder's syndrome is a benign entity with a favorable outcome and a self-limited course, although slight miosis and ptosis may remain.
Minton and Bounds reported five additional male cases belonging to Group II as defined by Boniuk and Schlezinger, all having an excellent prognosis, with pain subsiding within a few weeks (7). Two of their cases, though, had a diminished sensation of the cornea on the pain side.
Grimson and Thompson went one step further, proposing a subdivision into three groups, and added 41 cases of their own (8). Group I was identical to Boniuk's group I, i.e. “Ræder's syndrome proper”. Group II was cluster headache and Group III merely a painful postganglionic Horner's syndrome, although involvement of only the first branch of the trigeminal nerve was allowed in this group. Most patients belong to either of the two latter groups, which both have an excellent prognosis and are in no need of extensive diagnostic work-up. Group I, on the contrary, often harbors a malignant lesion and should be aggressively examined.
Mokri drew attention to the fact that spontaneous internal carotid artery dissection may be a frequent cause of painful postganglionic Horner's syndrome, i.e. Ræder's syndrome Group II (Boniuk) or III (Grimson). In a thoughtful and provocative contribution he discards the use of the eponym Ræder to other cases than the so-called Group I; in his view, inclusion of the other groups merely clouds diagnostic consideration (9, 10).
Watanabe et al. added neurosyphilis to the list of conditions that may cause a proper Ræder's syndrome (11). Their patient had involvement of the two upper divisions of the trigeminal nerve in addition to pain, ptosis and miosis, and became free of symptoms and signs within 2 weeks of the start of treatment with penicillin.
Ottar Sjaastad et al. elaborate their view in a comprehensive contribution (12). They propose a third way of dividing the syndrome; a modification of Boniuk and Schlezinger's and Grimson and Thompson's proposals. According to Sjaastad, Group IA is the Ræder's syndrome proper, with involvement of the trigeminal and other parasellar cranial nerves, while Group IB implies affection of the trigeminal nerve only and no other cranial nerves, admitting that involvement of the trigeminal will usually only be minor. Group II is a painful postganglionic Horner's syndrome identical to the group II of Boniuk and Schlezinger.
Sjaastad et al. describe six patients, five of them demonstrating the picture of Group IB. They elaborate the patterns of pupillary and sweating abnormalities, and convincingly demonstrate that both are of a postganglionic type. As for sweating, this implies that there is an area of “anhidrosis”, but that this area is restricted and localized to the medial supraorbital area only. Sweat glands in this area are most probably innervated via fibers from the internal carotid plexus. The outcome for their patients was good.
Sjaastad admits that accepting Group II patients as cases of Ræder's syndrome might create confusion versus cluster headache. However, the temporal pattern is very different, Ræder's syndrome running a self-limited course and very rarely recurring, and never with the clustering phenomenon. Moreover, cluster headache patients are mostly younger, Ræder's syndrome mainly afflicting middle-aged men. The pain is more excruciating in cluster headache.
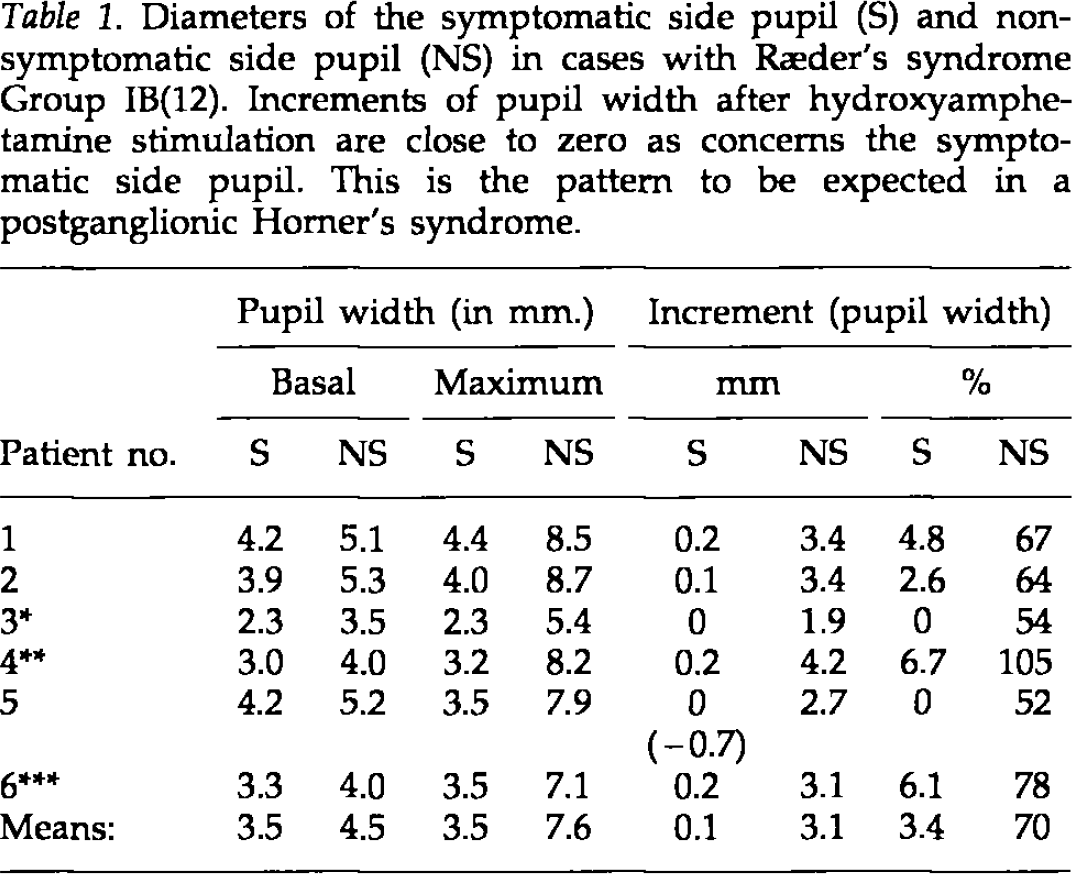
Furthermore, the pattern of oculosympathetic paresis evaluated in pharmacopupillary testing seems to be different in Ræder's syndrome versus cluster headache. The small pupil in Ræder's cases demonstrates no dilatation whatsoever on stimulation with hydroxyamphetamine eye drops, in this way behaving as a postganglionic deficit of other cause (Table 1). In cluster headache, on the other hand, the small pupil demonstrates some dilatation after hydroxyamphetamine stimulation, even in the patients with a basal miosis as pronounced as in Ræder's syndrome (Fig. 2). These patterns might reflect a more fundamental difference in the mechanism of the miosis in the two conditions.
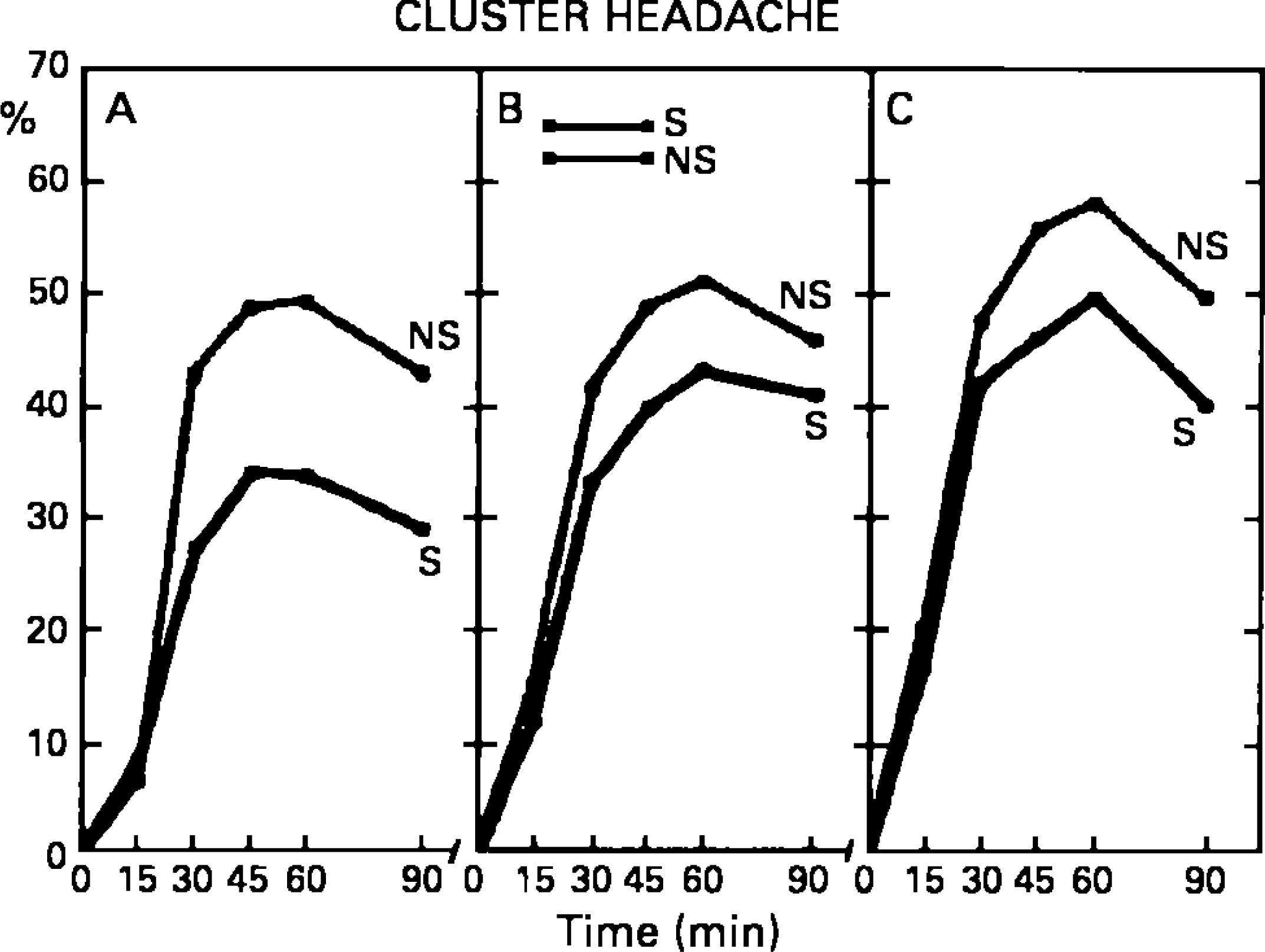
Dilatation of symptomatic side pupil (S) and nonsymptomatic side pupil (NS) in cluster headache patients after stimulation with hydroxyamphetamine eye drops. Relative dilatation in percent of basal diameters at various time intervals. Group 1 comprises patients with the more pronounced basal pupil asymmetry (>0.5 mm), approaching the asymmetry in cases with Ræder's syndrome. Note that even in this group, the symptomatic side dilatation amounts to 30% (from 13).
Diameters of the symptomatic side pupil (S) and non-symptomatic side pupil (NS) in cases with Ræder's syndrome Group IB(12). Increments of pupil width after hydroxyamphetamine stimulation are close to zero as concerns the symptomatic side pupil. This is the pattern to be expected in a postganglionic Horner's syndrome.
Tolosa-Hunt may be another diagnosis to be considered, but these patients rarely have an oculosympathetic paresis. While trigeminal involvement is the cardinal finding of Ræder, oculomotor involvement is the hallmark of Tolosa-Hunt syndrome. Furthermore, Tolosa-Hunt has no sex predominance, while Ræder's syndrome might be considered as a “super-male headache”, with an overwhelming majority of patients being males, in the same way as for SUNCT.
I would assume that this latter contribution is one expression of the current view. Ræder's syndrome proper might be subdivided into groups. Group IA, demanding affection of the trigeminal nerve and oculomotor nerves, carries a grave prognosis deserving a thorough diagnostic work-up. Group IB has no oculomotor involvement, and usually demonstrates a much better prognosis. Group II is merely a painful postganglionic Horner's syndrome, and demonstrates an excellent prognosis with the disease running a self-limited course. The latter two groups almost exclusively affect male patients, usually middle-aged. The tendency to recurrence is so small as almost to be neglected. The term “paratrigeminal” should be restricted to Ræder's syndrome Group I, possibly Group IA.