
Abstract
Objective
To investigate whether blockade of hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels modifies migraine induced by activation of vascular ATP-sensitive potassium (KATP) channels.
Methods
We conducted a single-centre, randomised, double-blind, placebo-controlled, two-way crossover study in adults with migraine without aura. On two separate days, participants received intravenous levcromakalim followed immediately by either oral ivabradine or placebo in a balanced order. The primary endpoint was the 12-h incidence of levcromakalim-induced migraine. Secondary endpoints included the area under the curve (AUC) for headache intensity and haemodynamic responses. Parallel preclinical experiments were performed in a validated mouse model using von Frey-based tactile sensitivity to assess whether ivabradine, given as pretreatment or as rescue medication, alters levcromakalim-induced hypersensitivity.
Results
Twenty seven of 31 individuals completed the human study and provided data for the final analysis. Ivabradine did not modify the incidence of levcromakalim-induced migraine (22 of 27 after ivabradine and 22 of 27 after placebo; P > 0.99) or the AUC for headache intensity (P = 0.11). Haemodynamic responses did not differ between study days. In mice, ivabradine at multiple doses neither prevented nor reversed tactile hypersensitivity induced by repeated levcromakalim administration.
Conclusions
HCN channel blockade does not influence migraine or nociceptive behaviour provoked by vascular KATP channel activation. These convergent human and preclinical findings indicate that HCN channels are not essential for the downstream transformation of vascular KATP channel activation into migraine pain and support a model in which migraine initiation arises from signalling at the vessel-to-neuron interface.
Trial registration
ClinicalTrials.gov; NCT04853797; Registered: 16-03-2021. Preclinical experiments were not preregistered beyond the animal ethical license (2017-15-0201-01358) from the Danish Animal Experiments Inspectorate.
This is a visual representation of the abstract.
Keywords
Introduction
Understanding how migraine pain is initiated remains a central challenge in headache research. 1 Human provocation studies have shown that intravenous levcromakalim, an ATP-sensitive potassium (KATP) channel opener with predominantly vascular action, 2 induces dilation of cranial arteries and migraine attacks in nearly all individuals with migraine without aura. 3 These observations demonstrate that activation of vascular KATP channels is sufficient to trigger migraine attacks.4,5 However, the molecular and cellular steps linking this vascular event to the activation of the trigeminovascular afferents are still debated.
Two mechanistic frameworks have been proposed (Figure 1). One suggests that migraine pain arises from direct activation of meningeal nociceptors (Figure 1A). In this neuronal model, KATP channel opening hyperpolarises peripheral afferents and recruits hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels, particularly HCN2 channels, which modulate excitability and nociception.6,7 Supporting this view, rodent studies have implicated HCN2 channels in migraine pain signalling. 7 The alternative vessel-to-neuron signalling hypothesis (Figure 1B) proposes that migraine is initiated within the meningeal vasculature. 8 Activation of vascular smooth-muscle KATP channels produces membrane hyperpolarisation, vasodilation, and potassium efflux into the perivascular space. The resulting increase in extracellular potassium depolarises nearby perivascular trigeminal afferents.1,8 This mechanism provides a biologically coherent pathway through which vascular events can activate nociceptive fibres and initiate migraine pain.
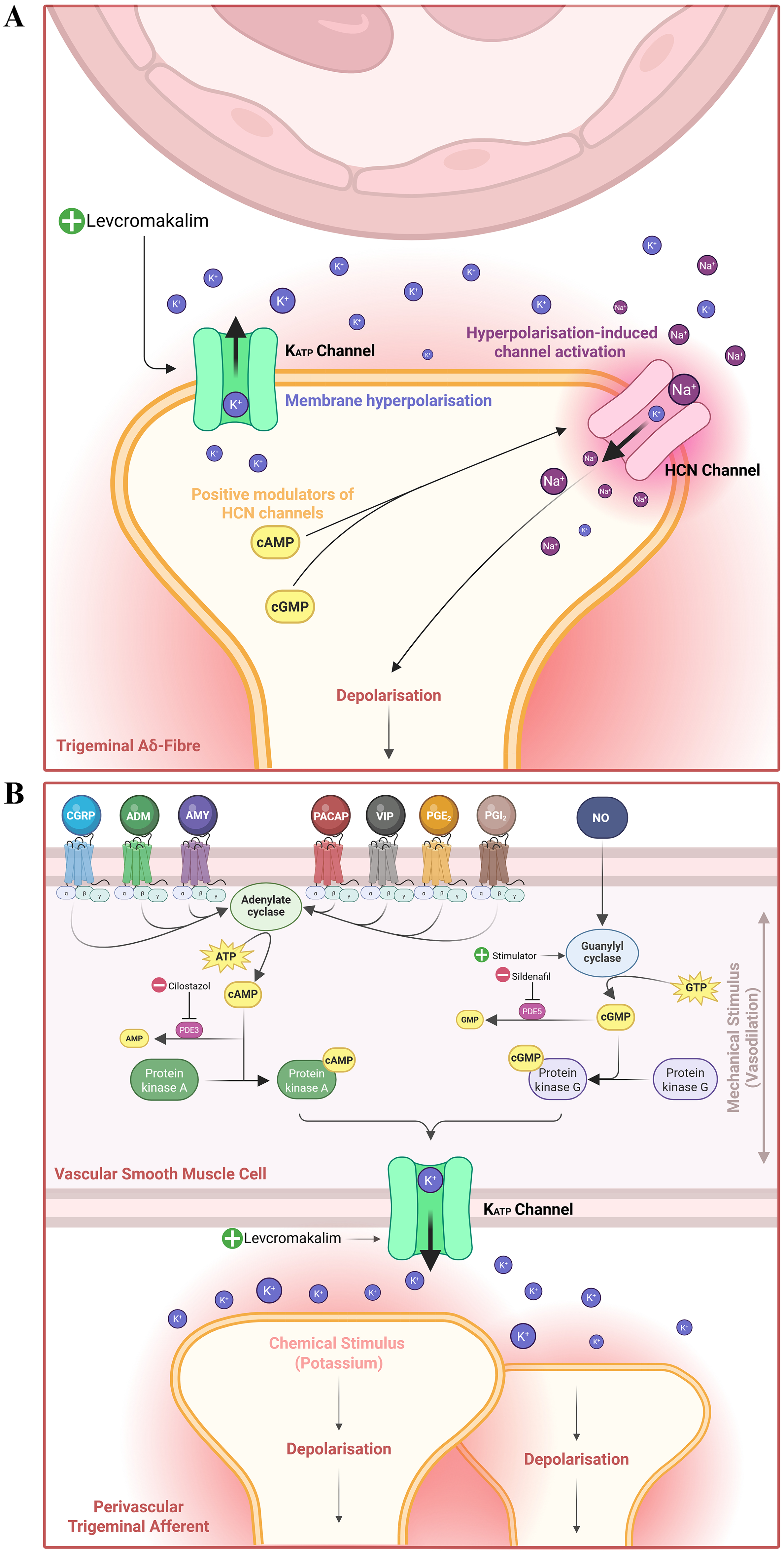
Neuronal hypothesis and vessel-to-neuron hypothesis. A: Neuronal hypothesis. Activation of ATP-sensitive potassium (KATP) channels hyperpolarises trigeminal Aδ fibres. Hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels open in response to hyperpolarisation and depolarise the fibre, thereby triggering nociceptive signalling. 6 B: Vessel-to-neuron hypothesis. Endogenous and exogenous migraine-triggering agents intersect within a shared signalling cascade in vascular smooth muscle cells, culminating with the activation of vascular KATP channels. Opening of these KATP channels produces membrane hyperpolarisation, vasodilation, and potassium efflux into the perivascular space, which depolarises nearby perivascular trigeminal afferents, generating nociceptive signals. 8
To discriminate experimentally between these models, we tested whether blockade of HCN channels with ivabradine (a clinically approved, heart rate lowering, non-selective HCN channel inhibitor with comparable efficacy across all four HCN isoforms) 9 modifies the migraine-inducing effect of levcromakalim. 3 We conducted a randomised, double-blind, placebo-controlled, two-way crossover study in individuals with migraine without aura, and complemented it with mechanistically aligned preclinical experiments using a validated mouse model of levcromakalim-induced tactile hypersensitivity. 10 This translational approach allowed us to evaluate whether activation of HCN channels is necessary for migraine initiation downstream of vascular KATP channel opening.
Materials and methods
The clinical protocol was based on the human migraine provocation model, 11 where the effect of early ivabradine treatment on levcromakalim-induced migraine was examined. Preceding this, a pilot study was carried out to assess safety and effects of ivabradine on levcromakalim-induced symptoms in healthy volunteers (only tension-type headache ≤ 5 days/month on average over the last year was allowed). All participants provided written informed consent prior to any study-related procedures. The study protocol was approved by the Regional Health Research Ethics Committee of the Capital Region of Denmark (H-20061329). The study was conducted at a single centre in Denmark in accordance with the Declaration of Helsinki with later revisions, registered with ClinicalTrials.gov (NCT04853797), and reported in accordance with the CONSORT guidelines. The preclinical protocol was based on an established mouse model of levcromakalim-induced migraine, 10 where the effect of ivabradine administered prior to and after levcromakalim administration was examined. The experiments were conducted under license number 2017-15-0201-01358 from the Danish Animal Experiments Inspectorate and performed and reported in accordance with the ARRIVE guidelines.
Clinical study design
In a randomised, double-blind, placebo-controlled, two-way crossover study, eligible adults diagnosed with migraine without aura 12 were allocated to receive an intravenous (IV) infusion of levcromakalim on two different study days, followed by oral administration of either ivabradine (2 × 7.5 mg tablets) or placebo (2×calcium tablets) in a random and balanced manner (Figures 2 and 3A). Participants received the other intervention on the alternate experimental day, facilitating exposure of all participants to both interventions. A minimum of one week separated the two experimental days, to ensure sufficient washout and avoid carry-over.

CONSORT 2010 Study Flow Diagram. Thirty-one of 368 potential participants were deemed eligible after screening and were enrolled and randomised. Of these, 27 participants completed the two study days, while four declined further participation after the first experimental day (one from the ivabradine-placebo sequence and three from the placebo-ivabradine sequence). Only data from the 27 participants who completed the study per protocol were included in the final analysis.

Study designs. A: Thirty-one participants diagnosed with migraine without aura were enrolled in a randomised, double-blind, placebo-controlled, two-way cross-over study. Four participants did not receive allocated intervention on the second experimental day (declined further participation). Twenty-seven participants completed both study days and received levcromakalim in combination with placebo and levcromakalim in combination with ivabradine on two different days with a minimum of seven days in between. B: Efficacy of intraperitoneal (IP) ivabradine or placebo (saline) administered as pretreatment at 10 mg/kg (cohort A) and 20 mg/kg (cohort B) 15 min prior to levcromakalim or vehicle on test days 1, 3, 5, and 7. Cohort A consisted of three parallel test groups, [SAL + VEH], [IVA + LEV] and [SAL + LEV], while cohort B consisted of four parallel groups, [SAL + VEH], [IVA + VEH], [SAL + LEV] and [IVA + LEV]. Von Frey test was performed to test for hypersensitivity prior to (basal measurement) and two hours after levcromakalim injection on every test day. Additionally, the efficacy of ivabradine as a rescue medication of established basal hypersensitivity was tested in Cohort B on day 9 (no additional levcromakalim injection given). After baseline measurements of hypersensitivity using von Frey testing on day 9, cohort B received IP ivabradine 5 mg/kg (time 0 min), followed by a repeat of withdrawal threshold tests thirty minutes later (time 30 min). At one hour (60 min) post-IP ivabradine 5 mg/kg, dosing was repeated and withdrawal thresholds were evaluated 30 min later (time 90 min). Following von Frey test on days 7 and 9, cohort B was tested on the rotarod to evaluate the effect of test substances on motor function. Half of cohort B was mildly sedated using midazolam as a positive control.
Participants
Eligible participants were adults aged 18-60 years with 1–5 migraine attacks without aura per month in the past year, as per the International Classification of Headache Disorders, 3rd edition (ICHD-3). 12 All participants were required to have a normal standard resting 12-lead electrocardiogram (ECG) at the screening visit with heart rate (HR) ≥ 60 bpm. Exclusion criteria included history of any other primary (except tension-type headache on ≤ 5 days/month on average over the last year) or secondary headache disorders, and daily intake of any medication other than oral contraceptives. The complete list of eligibility criteria is available at ClinicalTrials.gov (NCT04853797).
Randomisation and blinding
Simple, balanced randomisation was performed at a 1:1 ratio to allocate participants to the ivabradine-placebo or placebo-ivabradine sequence. Independent staff at the study site generated the randomisation code, which was stored in a lightproof envelope within a locked cabinet until study completion. Individual envelopes were prepared for each participant for potential emergency unblinding. Both participants and site investigators were blinded to treatment allocation.
Levcromakalim was synthesised and supplied in powder form by Tocris Bioscience (Bio-Techne) and subsequently sterilised and packaged by the Hospital Pharmacy of the Capital Region of Denmark. Ivabradine was procured as 7.5 mg tablets from KRKA, d.d., Novo mesto, Slovenia. Due to colour differences, independent staff unrelated to the study prepared 2 × 7.5 mg ivabradine tablets or size-matched 2×placebo (calcium) tablets in opaque containers to ensure blinding. Tablets were administered directly from the containers into the mouth of the participant to further preserve blinding. Participants were instructed to keep their eyes closed during administration and swallow the tablets as fast as possible with water to prevent accidental unblinding.
Procedures
Potential participants were identified by site investigators and invited for a screening visit, where eligibility was assessed based on predefined criteria. Participants underwent a semi-structured interview to capture clinical data, and a general physical and neurological examination. Vital signs, including HR and blood pressure, were measured, a 12-lead ECG was obtained, and females of childbearing potential underwent a urine pregnancy test. The participants were informed that levcromakalim might cause headache or migraine in some individuals; however, the expected timing and characteristics were withheld to avoid biasing responses. Participants were further informed that ivabradine might prevent or mitigate the effects of levcromakalim, though no information regarding potential mechanism(s) or site(s) of action were discussed.
On the two experimental days, participants arrived at the research lab at the same scheduled time between 08:00 and 13:00. Participants were instructed to refrain from all medications (except oral contraceptives) for at least 24 h or five drug half-lives before the experiment (whichever was longer), to avoid alcohol, caffeine, and tobacco for 12 h prior to the experiment, and to be fasting 2 h prior to experiment start; otherwise the session was rescheduled. Additional reasons for rescheduling participants included experiencing a headache within 24 h or a migraine attack within 48 h of experiment start. Females of childbearing potential were required to have a negative pregnancy test. A venous catheter was placed in the antecubital vein for drug administration, and participants were placed in a supine position in a controlled and quiet laboratory environment with dimmed lighting for at least 30 min before baseline measurements. These included headache assessment, vital signs, ECG, diameter of the left superficial temporal artery (STA), and middle cerebral artery blood flow velocity (VMCA). The participants then received a 20-min continuous IV infusion of levcromakalim (0.05 mg/min), followed immediately by oral administration of either ivabradine (2 × 7.5 mg tablets) or placebo (2×calcium tablets). Regular assessments of headache characteristics and associated symptoms, vital signs, ECG, diameter of the left STA and VMCA were conducted throughout the 150-min in-hospital observation period following the start of levcromakalim infusion. After completing the in-hospital monitoring, participants were discharged and instructed to complete a headache diary with hourly entries until 12 h post-levcromakalim infusion start (Figure 4). Participants were permitted to use rescue medication at any timepoint, if needed, to treat their headache or migraine.

Clinical experimental timeline. Overview of the clinical experimental timeline. Heart rate (HR), blood pressure (BP), electrocardiogram (ECG), superficial temporal artery (STA) diameter, and middle cerebral artery blood flow velocity (VMCA) were measured during the 2.5 h in-hospital observation period. Headache characteristics and associated symptoms were evaluated using a headache diary. Participants were instructed to complete hourly home registrations of the headache diary from discharge until 12 h after study start.
Headache and associated symptoms
A headache diary was used to record headache characteristics, associated symptoms, rescue medication use, and adverse events from baseline and every 10 min until 150 min post-levcromakalim infusion. Participants then completed hourly home entries until 12 h after the start of levcromakalim infusion. The timeframe was chosen for feasibility reasons and for close alignment with participants’ daily routines to maximise data collection.
The headache diary included information on headache features such as pain intensity (quantified using the numerical rating scale, NRS: 0 = no pain; 1 = changed but not painful sensation; 10 = worst imaginable pain), pain quality, pain location, aggravation of pain by routine physical activity, presence of migraine-associated symptoms (nausea, photophobia, phonophobia), resemblance of headache to usual attacks, and other patient-reported symptoms (heat sensations, flushing, palpitations etc.).
Experimental criteria for migraine attacks
The criteria for defining an experimentally induced migraine attack within the 12-h observation period were:
11
Headache fulfilling ICHD-3 criteria C and D for migraine without aura
12
and/or Headache with features mimicking the participant's usual migraine and treated with their usual acute migraine medication.
Haemodynamic parameters
Vital signs, including HR and blood pressure, were monitored every 10 min from baseline until 150 min after levcromakalim infusion using a bedside monitor. Mean arterial pressure (MAP) was calculated based on measured systolic and diastolic blood pressures. ECG was monitored continuously and printed at baseline and every 10 min from 50 min after levcromakalim infusion start until end of in-hospital observation period. VMCA was measured bilaterally using transcranial doppler with 2-MHz handheld probes (TCD; Doppler BoxX with simultaneous open-mask no respiratory resistance CO2 recordings using ProPaq Encore, Welch Allyn Protocol; Multi-Dop X with integrated CO2 measuring module). Diameter of the frontal branch of the left STA was measured using high-frequency ultrasound (Dermascan C, Cortex Technology, Hadsund, Denmark; 20 MHz, bandwidth 5 MHz). Both the VMCA and diameter of STA were recorded at baseline and every 20 min until 150 min after start of levcromakalim infusion. For accuracy and reliability, we calculated the mean of four measurements of VMCA and STA diameter per timepoint.
Preclinical study design
Mouse model and behavioural testing
We utilised a mouse model of levcromakalim-induced migraine to investigate the effect of ivabradine as pretreatment prior to injection of levcromakalim and as rescue medication for established levcromakalim-induced basal hypersensitivity. 10 In brief, cutaneous tactile hypersensitivity to stimulation with von Frey filaments was induced in wild-type mice by repeated systemic intraperitoneal (IP) administration of levcromakalim (1 mg/kg). Levcromakalim was injected every other day, and tactile sensitivity was evaluated prior to injection (basal response) and 2 h after injection. As reported previously, mice generally develop tactile hypersensitivity at the 2-h time point after 1–2 injections of levcromakalim, and at the basal time point following 2–3 injections, corresponding to test days 1–3 and 3–5, respectively. Tactile hypersensitivity was evaluated on the plantar surface of the left hind paw using von Frey filaments (0.008–2 g, excluding 1.4 g, Ugo Basile) applied in the up-down paradigm by an experimenter blinded to treatment groups. 13 Fifty percent withdrawal thresholds were calculated using the online calculator described by Christensen et al., with exact delta values and filament force used as settings. 14
Mouse motor function was assessed immediately after von Frey testing on selected days to ensure that tactile sensitivity measurements were not influenced by drug-induced side effects, such as mild sedation. Motor performance was evaluated using the Rotarod Advanced (IITC Life Science). The mice were given a single attempt on the rotarod with no prior training. Rotation started at 0 rpm and gradually accelerated to 30 rpm over 45 s. The test was terminated after a maximum of 150 s. Midazolam (2 mg/kg, IP) was administered afterwards as a positive control.
Animals
Two cohorts of mice (A and B) were included in the experiments, comprising a total of 84 C57BL/6J BomTac (Taconic, Denmark) male and female mice, aged 8–10 weeks. Mice were group-housed in a temperature (21–23 °C) and humidity (55–65%) controlled facility with free access to food and water and a 12-h light cycle (lights on at 07:00). Multiple types of shelters and nesting materials were provided for enrichment purposes. Further details are provided in Christensen et al. 10 Upon arrival, mice were acclimatised for one week before initiation of experiments. A priori humane endpoints and exclusion criteria included weight loss (≥ 10%), signs of illness, wounds, or non-compliance with behavioural testing; however, no animals met these criteria.
Experimental design
In cohort A, we evaluated the efficacy of IP ivabradine at 10 mg/kg.
15
The study design included three parallel test groups; [SAL + VEH], [SAL + LEV] and [IVA + LEV] corresponding to saline followed by vehicle, saline followed by levcromakalim, and ivabradine followed by levcromakalim. Ivabradine or saline was administered 15 min prior to levcromakalim or vehicle on test days 1, 3, 5, and 7. Von Frey testing was performed prior to and 2 h after levcromakalim injection on every test day (Figure 3B
Cohort B initially followed the same protocol as cohort A; however, the ivabradine dose was increased to 20 mg/kg IP,7,16 and the design included four parallel experimental groups instead of three, with the addition of [IVA + VEH] (ivabradine followed by vehicle). Additionally, cohort B was used to assess the efficacy of ivabradine in reversing established basal hypersensitivity on day 9 (no additional levcromakalim was administered). After the basal measurement, mice received ivabradine (5 mg/kg, IP) at time 0 min, and withdrawal thresholds were evaluated 30 min later. Sixty minutes after the first ivabradine dose, dosing was repeated, and withdrawal thresholds were evaluated 30 min later (time 90 min) (Figure 3B). 7
Both mouse cohorts were allocated to treatment groups with stratification by sex, home cage, and baseline 50% withdrawal thresholds. Following von Frey testing on days 7 and 9, all mice in cohort B were assessed twice on the rotarod: first to evaluate the effects of the test substances, and second as a positive control, in which mild sedation was induced by midazolam in half of the mice (Figure 3B).
Study compounds
Levcromakalim (Tocris Bioscience, Bio-Techne), was dissolved in saline with 2% DMSO. Ivabradine HCl (Tocris Bioscience, Bio-Techne), and midazolam (Hameln Pharma GMBH, Germany, via the Hospital Pharmacy of the Capital Region of Denmark) was dissolved in saline. All compounds and their corresponding vehicles were injected in a volume of 10 ml/kg.
Statistical analysis
All analyses of human data were performed in GraphPad Prism for Windows (version 8.4.2.679) without adjustment for multiple comparisons. Sample size calculation was performed in R (version 4.1.2). Descriptive statistics are presented as frequencies and percentages for categorical variables, and as means ± standard deviations (SD) or medians with ranges for continuous variables, depending on normality. Headache-related data are consistently reported as medians with ranges. Normality was assessed using the Shapiro-Wilk test and QQ-plot inspection.
The primary endpoint of the human experimental study was the incidence of migraine attacks during the 12-h observational period following levcromakalim administration. Sample size was estimated using a two-sided McNemar's test for paired proportions (package: MESS) and informed by migraine induction- and interventional efficacy data from previous studies,1,3,4,17–19 and a hypothesised, meaningful ivabradine effect size. We expected 10% of participants to report migraine attacks only after ivabradine and 50% only after placebo. With 80% power and a 5% significance level, a sample size of minimum 27 participants was required. McNemar's test was used to analyse the primary endpoint results, while period and carry-over effects on the primary outcome were evaluated with Fisher's exact test. 20
Secondary endpoints included headache incidence, area under the curve (AUC) for headache intensity scores, and incidence of adverse events. Other endpoints included AUC for changes in STA diameter, VMCA, MAP, and HR during the 150-min in-hospital. Differences in headache induction and adverse events were analysed using McNemar's test. AUC values were calculated with the trapezium rule, and differences in AUC for headache intensity scores were assessed with the Wilcoxon signed-rank test, while AUC for changes in STA diameter, VMCA, MAP, and HR were analysed with paired t-tests. All statistical tests were two-tailed, and significance was set at P < 0.05.
Data from the preclinical experiments were analysed and visualised using GraphPad Prism, version 10.4.1. Group sizes of 12 were determined a priori based on prior experience with the model and tactile allodynia as the primary endpoint, ensuring adequate power to detect intermediate antagonist effect sizes in a four-arm repeated-measures design. Raw 50% withdrawal thresholds were square-root (SQRT) transformed to improve normality and meet parametric assumptions. Transformed von Frey data are shown as individual data points and/or mean ± standard error of the mean (SEM) for each test day. Data were analysed using a repeated-measures mixed-effects model with Geisser–Greenhouse correction and Dunnett's multiple comparisons test, with the [SAL + LEV] group as the reference. Mean differences with 95% confidence intervals (CIs) are reported in the Results for key comparisons, with full statistics provided in Online Supplementary Tables S1a-d. Model residuals were assessed via QQ-plots to confirm assumption validity. Rotarod data are shown as individual data points with medians and analysed using the Kruskal–Wallis test followed by Dunn's multiple comparisons test, comparing each test group to [SAL + VEH] and midazolam to saline in the positive control experiments.
Data availability
Access to deidentified study data may be granted by the corresponding author upon reasonable request.
Results
Clinical results: The human migraine model
Participants
Six healthy volunteers were enrolled in the pilot study between May 2021 and October 2021 and completed both study days (data not shown). A total of 31 participants were enrolled in the main study between October 2022 and July 2025 and underwent randomisation, of which 27 completed both experimental study days and provided data for the final analysis (Figures 2 and 3). Demographic and clinical characteristics at screening for all completed participants are shown in Table 1.
Demographic and clinical characteristics of the human study population at screening.

Screening characteristics of all who completed both study days (n = 27) and by treatment sequence.
Migraine
Twenty-two of 27 (81%) participants reported levcromakalim-induced migraine during the 12-h observational period on both study days, with three (11%) reporting migraine exclusively after ivabradine and three (11%) exclusively after placebo (P > 0.99; Table 2). Nineteen of 27 (70%) reported migraine on both days, and two (7%) on neither. Median time to migraine onset was 180 min (range: 40–360 min) after ivabradine and 240 min (40–540 min) after placebo. No significant period or carry-over effects for migraine induction were observed between the two study days (P > 0.99 and P = 0.48, respectively).
Clinical characteristics of spontaneous and provoked (0–12 h observational period) headache and associated symptoms in participants with migraine without aura.

Localisation/intensity/quality (throb = throbbing; pres = pressing)/aggravation or avoidance of physical activity (by cough during in-hospital phase and by movement during out-hospital phase).
Nausea / photophobia / phonophobia.
Migraine-like attacks are defined according to criteria described in methods.
Pain freedom or pain relief (≥ 50% decrease in intensity) within 2 h.
Abbreviations: ODT, oral disintegrating tablet; TAB, tablet; TBEF, effervescent tablet; SC, subcutaneous; IV, intravenous.
Headache
Twenty-five of 27 (93%) participants reported levcromakalim-induced headache of any intensity after ivabradine compared with 24 of 27 (89%) after placebo (P > 0.99; Table 2). Of these, two of 27 (7%) had headache exclusively after ivabradine, one of 27 (4%) had headache exclusively after placebo and 23 of 27 (85%) had headache on both study days. One participant (4%) had no headache on either day. The AUC0–720 min values for headache intensity scores did not differ between the two experimental days (P = 0.11; Figure 5). Median time to headache onset was 20 min (range: 10–240 min) after ivabradine and 30 min (range: 10–420 min) after placebo, with median peak headache intensity being 4 (range after ivabradine: 0–10 vs. range after placebo: 0–9) and median time to peak headache intensity being 240 min on both days (range after ivabradine: 30–540 min vs. range after placebo: 30–540 min).

Headache intensity. Median headache intensity (bold lines) is presented on the numerical rating scale (NRS) after levcromakalim administration at T0 min followed by oral ivabradine or placebo, respectively, at T20 min. Individual headache scores are shown as transparent lines. Participants were observed for 12 h in total.
Haemodynamic parameters
No differences were observed between the two study days with respect to AUC0−150 min for STA diameter changes (P = 0.46), VMCA changes (P = 0.81), MAP changes (P = 0.52), and HR changes (P = 0.44). Mean percentage changes of STA diameter and VMCA are shown in Figure 6, and mean percentage changes of MAP and HR are shown in Figure 7. Absolute changes in STA diameter, VMCA, MAP and HR are shown in Online Supplemental Figures S1 and S2. Baseline haemodynamic values prior to administration of levcromakalim are shown in Online Supplementary Table S2.
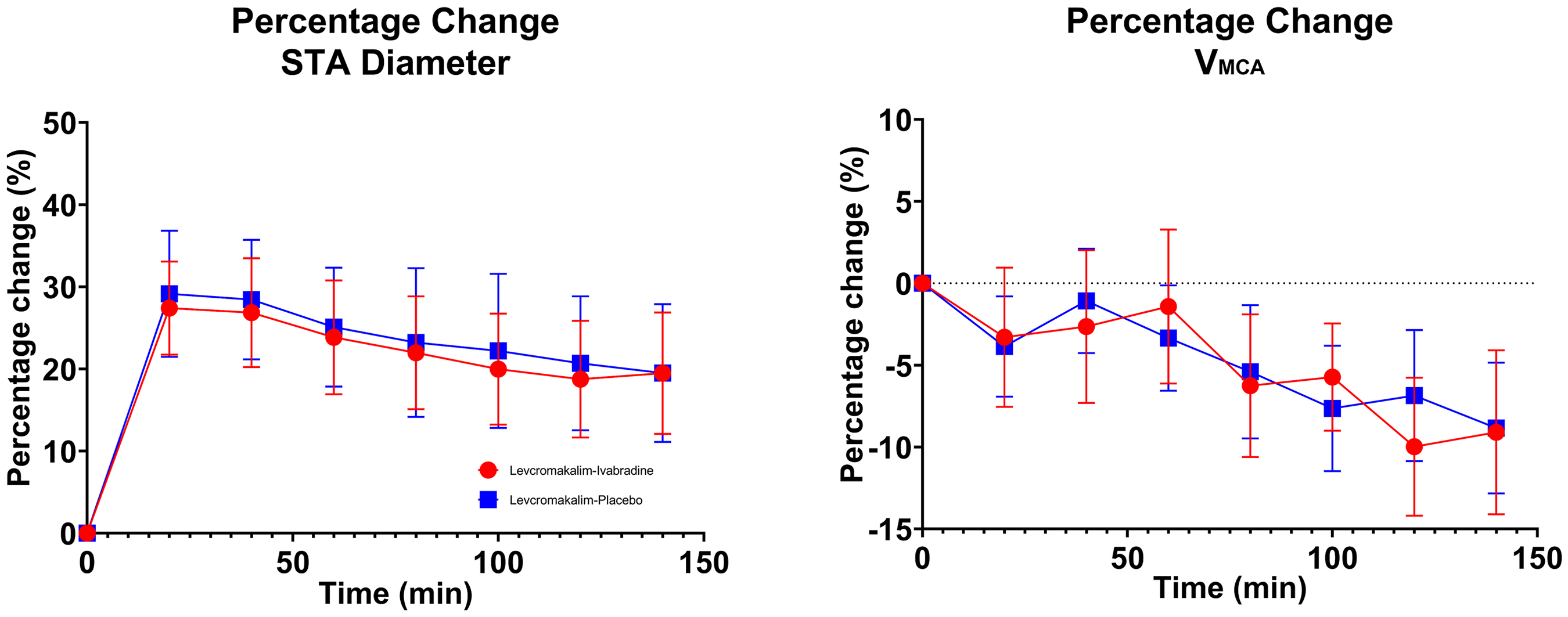
Percentage change in superficial temporal artery (STA) diameter and middle cerebral artery blood flow velocity (VMCA). Mean percentage change in diameter of superficial temporal artery (STA) and mean percentage change in middle cerebral artery blood flow velocity (VMCA) from baseline on ivabradine day (red) and placebo day (blue) until 150 min after start of levcromakalim-infusion.

Percentage change in mean arterial pressure (MAP) and heart rate (HR). Mean percentage change in mean arterial blood pressure (MAP) and mean percentage change in heart rate (HR) from baseline on ivabradine day (red) and placebo day (blue) until 150 min after start of levcromakalim-infusion.
Adverse events and rescue medication use
Adverse events occurred in all 27 (100%) participants on both study days (McNemar's 2-sided test for paired data not applicable). The most common adverse events reported were flushing, warmth and palpitations (Table 3). One participant reported short-lasting orthostatic hypotension not requiring hospitalisation in the out-of-hospital phase after ivabradine. No serious adverse events occurred. Rescue medication use was reported by 23 of 27 (85%) participants after ivabradine and 22 of 27 (81%) after placebo. Median time to rescue medication was 240 min on both study days (range: 140–360 min after ivabradine vs. range: 120–448 min after placebo).
Most common adverse events reported.

Adverse events occurring within the 12-h observational study-period after ivabradine and placebo, respectively.
Preclinical results: Mouse model of levcromakalim-induced migraine
Ivabradine did not prevent levcromakalim-induced mechanical hypersensitivity
In cohort A (N = 36), ivabradine (10 mg/kg, IP) was administered 15 min prior to levcromakalim on test days 1, 3, 5 and 7 in a three-arm design including test-, positive control, and negative control groups (Figure 8A). Overall, there were significant effects of time [F(2.796, 92.26) = 7.936, P = 0.0001] and treatment [F(2, 33) = 13.01, P < 0.0001], but no significant interaction [F(6, 99) = 1.046, P = 0.4]. Dunnett's post hoc test showed that mild hypersensitivity developed in the levcromakalim [SAL + LEV] group compared with the negative control [SAL + VEH] group, but this was not prevented by ivabradine [IVA + LEV] on any test day. The mean differences [95% CI] between [IVA + LEV] and [SAL + LEV] were: Day 1: −0.11 [−0.37 to 0.16], P = 0.53; Day 3, −0.03 [−0.31 to 0.26], P = 0.97; Day 5, 0.02 [−0.19 to 0.21], P = 0.98; and Day 7, −0.14 [−0.41 to 0.13], P = 0.37.

Effect of ivabradine on levcromakalim-induced tactile hypersensitivity in a mouse model. Group size = 12 per group. SAL = saline, VEH = vehicle, LEV = levcromakalim, IVA = ivabradine, MIDA = midazolam. Significance levels (adjusted p-values): p < 0.05, *p < 0.01, **p < 0.001, ***p < 0.0001. A: Cohort A. Left: baseline tactile sensitivity prior to group allocation; points represent individual mice, with mean ± standard error of the mean (SEM). Right: tactile sensitivity 2 h after injection of test substances; data are mean ± SEM. Analysis: repeated-measures mixed-effects model with Dunnett's post hoc comparison with [SAL + LEV] as reference. B: Cohort B. Left: baseline tactile sensitivity prior to group allocation; points represent individual mice, with mean ± SEM. Right: tactile sensitivity 2 h after injection of test substances; data are mean ± SEM. Analysis: repeated-measures mixed-effects model with Dunnett's post hoc comparison with [SAL + LEV] as reference. C: Cohort B, day 9. Basal hypersensitivity before and after ivabradine injection at 0 and 60 min; mice did not receive levcromakalim on this day. Data points represent individual mice, with mean ± SEM. Analysis: repeated-measures mixed-effects model with Dunnett's post hoc comparison with [SAL + LEV] as reference. D: Cohort B: Motor performance assessed after von Frey testing on days 7 (left) and 9 (right). Data points represent individual mice with medians. Analysis: Kruskal–Wallis test with Dunn's multiple comparisons with [SAL + VEH] as reference and midazolam with saline in the positive control experiments.
The experiment was repeated with a higher dose of ivabradine (20 mg/kg, IP) in a separate cohort (B) of 48 mice (Figure 8B). This design included an additional group [IVA + VEH] to evaluate potential side effects of the higher ivabradine dose. Overall, there were significant effects of time [F(2.775, 122.1) = 15.61, P < 0.0001] and treatment [F(3, 44) = 18.41, P < 0.0001], but no significant interaction [F(9, 132) = 1.578, P = 0.13]. Post hoc analysis showed that levcromakalim [SAL + LEV] induced hypersensitivity on all test days compared with the negative control [SAL + VEH] group (P = 0.014–0.0001). The [IVA + VEH] group, confirmed that ivabradine had no effect on withdrawal thresholds. The higher ivabradine dose (20 mg/kg, IP) in the [IVA + LEV] group likewise failed to prevent levcromakalim-induced hypersensitivity. The mean differences [95% CI] between [IVA + LEV] and [SAL + LEV] were as follows: Day 1, −0.08 [−0.46 to 0.31], P = 0.92; Day 3, −0.04 [−0.36 to 0.28], P = 0.98; Day 5, 0.11 [−0.14 to 0.36], P = 0.57; and Day 7, 0.13 [−0.15 to 0.41], P = 0.49.
Ivabradine did not reverse established mechanical hypersensitivity
Next, we tested whether ivabradine could reverse established levcromakalim-induced hypersensitivity (Figure 8C). Cohort B (N = 48) continued to test day 9, where they only received ivabradine (2 × 5 mg/kg, IP, one hour apart) following an initial assessment of basal tactile sensitivity. Von Frey testing was repeated 30 min after each ivabradine injection. Analysis revealed a significant effect of treatment [F(3, 44) = 35.69, P < 0.0001], but no significant effects of time [F(1.988), 87.48) = 2.958, P ampa#thinsp;0.057] or treatment×time interaction [F(6, 88) = 1.094, P = 0.37]. Post hoc testing confirmed basal tactile hypersensitivity in both groups that received levcromakalim on days 1–7 compared with the two vehicle-treated groups. No difference was observed between the ivabradine and saline-treated groups after the first ivabradine injection (mean difference: 0.07 [−0.14 to 0.29], P = 0.73). Similarly, no difference was observed 30 min after the second injection (mean difference: 0.14 [−0.06 to 0.34], P = 0.22).
Motor function
Motor function in cohort B was assessed after von Frey testing on days 7 and 9 (Figure 8D). No significant adverse effects were observed with any combination of ivabradine (20 mg/kg or 2 × 5 mg/kg), levcromakalim, or vehicle. Median rotarod times on day 7 were 150 s for [SAL + VEH], [IVA + VEH], and [SAL + LEV], and 87.5 s for [IVA + LEV] (mean rank difference 13.64, P = 0.74). Medians were 98, 66.5, 115, and 74.5 s, respectively, on day 9. Midazolam-sedated mice failed the task on both test days (day 7: 39 vs 150 s, mean rank difference 42.3, P < 0.0001; day 9: 34.5 vs 104.5 s, mean rank difference 40.3, P < 0.0001).
Discussion
The clarification of mechanisms by which migraine pain is initiated remains essential for understanding the interaction between vascular and neuronal elements within the meninges. This study examined whether inhibition of HCN channels modifies migraine attacks induced by vascular KATP channel activation in humans and whether it alters levcromakalim induced tactile hypersensitivity in mice. Across both experimental platforms, ivabradine produced no detectable effect on any migraine relevant outcomes, suggesting that HCN channel activity is not required for KATP channel-triggered migraine. These findings support a model in which migraine initiation arises from vascular-to-neuronal signalling 8 rather than from direct HCN channel-dependent neuronal mechanisms.
Interpretation of clinical findings
Consistent with previous reports, intravenous levcromakalim reliably provoked migraine attacks,3–5 yet ivabradine neither prevented nor attenuated these responses. This absence of effect aligns with the selectivity of levcromakalim for the vascular KATP channel Kir6.1/SUR2B subtype, 2 and indicates that acute HCN channel signalling alone is not required to transform vascular hyperpolarisation into migraine pain. Additional observations from related studies, including the lack of migraine induction by neuronal KATP channel activation 21 and the absence of nociceptive responses after extracranial levcromakalim administration, 22 further support the conclusion that the initiating events occur within the intracranial vasculature rather than in peripheral neurons. 8 The absence of ivabradine effects on headache intensity, onset, and haemodynamic variables suggests that HCN channels are unlikely to play a dominant role in early trigeminovascular responses downstream of vascular smooth muscle activation.
Interpretation of preclinical findings
Consistent with the human data, the preclinical mouse experiments closely mirrored the clinical findings: ivabradine failed to prevent or reverse levcromakalim-induced mechanical hypersensitivity in mice at doses previously demonstrated to affect pain behaviour in other preclinical pain- and migraine models.7,15,16 A single dose of ivabradine at 5 mg/kg IP, compared with the 10 and 20 mg/kg doses used in the present study, reversed mechanical thresholds to baseline in a mouse model of glyceryl trinitrate-induced migraine, with similar effects observed following targeted genetic deletion of HCN2 channels from voltage-gated sodium channel subunit alpha (NaV1.8)-expressing peripheral nociceptive neurons. 7 This further argues against a role for HCN channels in mediating the nociceptive responses downstream of vascular KATP channel activation. Prior work demonstrated that pharmacological inhibition10,23,24 or conditional loss-of-function of the vascular smooth-muscle Kir6.1 subunit 25 abolished migraine relevant hypersensitivity in animal models. Additionally, given that Kir6.1/SUR2B KATP channels are predominantly expressed in vascular smooth muscle and minimally in trigeminal ganglia, 25 the absence of ivabradine effect is consistent with a mechanism in which vascular activation and subsequent depolarisation of perivascular afferents constitute the key pathway, 8 independent of the HCN channel-driven changes in neuronal excitability described in other pain studies.15,26–32
Integrative mechanistic perspective
Taken together, these findings experimentally differentiate two mechanistic possibilities: one in which migraine depends on HCN channel-mediated neuronal processes 6 and another in which vascular events trigger afferent activation at the vessel-to-neuron interface. 8 The consistent lack of effect across species supports the latter, 8 suggesting that vascular hyperpolarisation and associated increases in extracellular potassium levels are sufficient to activate perivascular trigeminal fibres without an obligatory requirement for secondary HCN channel-mediated amplification. These findings underscore the importance of the meningeal microenvironment, where vascular and neuronal elements interact closely and illustrate how negative mechanistic results can refine conceptual models by excluding pathways that do not contribute to migraine initiation.
Relation to broader provocation pathways
Multiple migraine triggers converge on cyclic nucleotide signalling in vascular smooth muscle, with subsequent KATP channel activation representing a final common effector in this cascade.1,8,33–35 The present results suggest that this convergence can occur independently of downstream HCN channel activity, and that the vascular component of the trigeminovascular system may be sufficient to initiate headache. 8 While our findings argue against a critical downstream role of HCN channels in KATP channel-mediated migraine, a modulatory contribution cannot be excluded. Consistent with this, a single 15 mg dose of ivabradine had no effect in a human capsaicin pain model, 36 while two repeated-dosing studies, one open-label with seven participants 37 and another using a capsaicin model of pain, 38 were negative on primary pain outcomes, with isolated exploratory signals of possible HCN channel-related pain modulation. The agreement between human and animal findings indicates that vessel-to-neuron coupling may represent a conserved mechanism through which diverse upstream signals initiate migraine. 8
Strengths and limitations
Major strengths include the use of a validated human provocation model3,11 with a rigorous randomised crossover design, and parallel, mechanistically aligned, preclinical experiments based on an established mouse model of levcromakalim-induced migraine. 10 The timing and dosing of ivabradine specifically tested acute HCN channel involvement during migraine initiation, reflecting levcromakalim-mediated migraine dynamics, 3 and providing the most relevant window to assess its mechanistic role in migraine pathophysiology. The animal experiments used dosing paradigms previously shown to be behaviourally active,7,15,16 utilised different batches of ivabradine to ensure drug quality, included motor assessments to exclude confounding sedation, and tested both prevention and reversal of tactile hypersensitivity. Limitations include ivabradine's non-isoform-selective inhibition of all four HCN subtypes9, potential interspecies differences in tissue penetration, and anatomical variation in channel expression. Oral administration of ivabradine reflects standard clinical practice and should provide adequate exposure; the single 15 mg dose used here corresponds to the maximum licensed daily posology (maximum 7.5 mg twice daily) and has been shown to achieve clinically relevant plasma concentrations and robust HCN inhibition in humans.39–41 Intravenous dosing would have been preferable to further standardise exposure.39,41 The absence of a clear heart rate reduction following ivabradine intervention likely reflects opposing haemodynamic effects of levcromakalim-induced vasodilation and reflex sympathetic activation, rather than reduced ivabradine activity. Given the consistent clinical and preclinical results, further dose escalation is unlikely to alter outcomes and may be limited by tolerability. Possible long-term modulatory effects of sustained HCN channel inhibition in humans cannot be excluded based on this study. Despite these considerations, the convergence of evidence across two experimental systems strengthens the conclusion that HCN channels do not mediate KATP driven migraine.6,8
Conclusions
Blockade of HCN channels with ivabradine did not influence migraine attacks or nociceptive mouse behaviour induced by activation of vascular KATP channels. These findings argue against an acute critical role for HCN channels as key downstream mediators of KATP-triggered migraine and support a model in which migraine pain is initiated through coordinated signalling at the vessel-to-neuron interface within the meninges. Future therapeutic interventions might focus on modulating vascular-to-neuronal communication, stabilising extra-cellular ionic gradients, or regulating the release of mediators from perivascular afferents. Approaches directed at this interface may offer more precise modulation of migraine initiation than strategies aimed solely at neuronal excitability or vascular tone.
HCN channel blockade with ivabradine failed to modify migraine induced by vascular KATP channel activation in humans and did not affect levcromakalim-induced hypersensitivity in mice. The convergent clinical and preclinical data exclude HCN channels as essential downstream mediators of KATP-triggered migraine. These findings support vascular to neuronal signalling at the vessel-to-neuron interface as a key initiating mechanism in migraine pathophysiology.
Supplemental Material
sj-docx-1-cep-10.1177_03331024261431770 - Supplemental material for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation
Supplemental material, sj-docx-1-cep-10.1177_03331024261431770 for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation by Zixuan Alice Zhuang, Janu Thuraiaiyah, Lili Kokoti, Rogelio Dominguez Moreno, Emil Gozalov, Zahra Hakimzadeh, Mohammad Al-Mahdi Al-Karagholi, Amalie Clement, David M. Kristensen, Jawdat Abdulla, Sarah Louise Christensen and Messoud Ashina in Cephalalgia
Supplemental Material
sj-docx-2-cep-10.1177_03331024261431770 - Supplemental material for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation
Supplemental material, sj-docx-2-cep-10.1177_03331024261431770 for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation by Zixuan Alice Zhuang, Janu Thuraiaiyah, Lili Kokoti, Rogelio Dominguez Moreno, Emil Gozalov, Zahra Hakimzadeh, Mohammad Al-Mahdi Al-Karagholi, Amalie Clement, David M. Kristensen, Jawdat Abdulla, Sarah Louise Christensen and Messoud Ashina in Cephalalgia
Supplemental Material
sj-docx-3-cep-10.1177_03331024261431770 - Supplemental material for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation
Supplemental material, sj-docx-3-cep-10.1177_03331024261431770 for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation by Zixuan Alice Zhuang, Janu Thuraiaiyah, Lili Kokoti, Rogelio Dominguez Moreno, Emil Gozalov, Zahra Hakimzadeh, Mohammad Al-Mahdi Al-Karagholi, Amalie Clement, David M. Kristensen, Jawdat Abdulla, Sarah Louise Christensen and Messoud Ashina in Cephalalgia
Supplemental Material
sj-docx-4-cep-10.1177_03331024261431770 - Supplemental material for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation
Supplemental material, sj-docx-4-cep-10.1177_03331024261431770 for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation by Zixuan Alice Zhuang, Janu Thuraiaiyah, Lili Kokoti, Rogelio Dominguez Moreno, Emil Gozalov, Zahra Hakimzadeh, Mohammad Al-Mahdi Al-Karagholi, Amalie Clement, David M. Kristensen, Jawdat Abdulla, Sarah Louise Christensen and Messoud Ashina in Cephalalgia
Supplemental Material
sj-docx-5-cep-10.1177_03331024261431770 - Supplemental material for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation
Supplemental material, sj-docx-5-cep-10.1177_03331024261431770 for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation by Zixuan Alice Zhuang, Janu Thuraiaiyah, Lili Kokoti, Rogelio Dominguez Moreno, Emil Gozalov, Zahra Hakimzadeh, Mohammad Al-Mahdi Al-Karagholi, Amalie Clement, David M. Kristensen, Jawdat Abdulla, Sarah Louise Christensen and Messoud Ashina in Cephalalgia
Supplemental Material
sj-docx-6-cep-10.1177_03331024261431770 - Supplemental material for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation
Supplemental material, sj-docx-6-cep-10.1177_03331024261431770 for Migraine induced by vascular KATP channel activation is independent of HCN channel activity: A randomised controlled trial with translational validation by Zixuan Alice Zhuang, Janu Thuraiaiyah, Lili Kokoti, Rogelio Dominguez Moreno, Emil Gozalov, Zahra Hakimzadeh, Mohammad Al-Mahdi Al-Karagholi, Amalie Clement, David M. Kristensen, Jawdat Abdulla, Sarah Louise Christensen and Messoud Ashina in Cephalalgia
Footnotes
Acknowledgements
The authors thank all participants for their invaluable contribution to this study, as well as all involved site personnel for their help in conducting the study. Figures 1 (Created in BioRender. Zhuang, Z. (2026) https://BioRender.com/q975w9v)![]() ) were created with Biorender.com.
) were created with Biorender.com.
ORCID iDs
Ethical considerations
The clinical study protocol was reviewed and approved by the Regional Health Research Ethics Committee of the Capital Region of Denmark (H-20061329). Animal experiments were conducted under license number 2017-15-0201-01358 from the Danish Animal Experiments Inspectorate.
Consent to participate
Written informed consent was obtained from all participants prior to enrolment, ensuring that they were fully informed and made aware of study assessments and procedures, and potential risks.
Author contributions
The study was conceived, initiated, and sponsored by MA. ZAZ, MMK and MA contributed to the clinical study design; protocol development; participant enrolment; data acquisition, data processing, data analysis, statistics, and interpretation; and drafting and revision of the article. JA contributed to study design; protocol development; and critical review of the article. JT contributed to participant enrolment; data acquisition, data processing, data analysis, and interpretation; and critical review of the article. LK, RDM, EG, and ZH contributed to participant enrolment; data acquisition and processing; and critical review of the article.
SLC, AC, and DMK contributed to design of the animal experiments; protocol development; data acquisition, data processing, data analysis, statistics, and interpretation; and drafting and revision of the article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by the Danish National Research Foundation (DNRF188; principal investigator, MA) and the Lundbeck Foundation Professor Grant (R310-2018–3711). The funding source was not involved in the design or conduct of the study; in the collection, analysis, or interpretation of the data; or in the preparation, review, or approval of the manuscript. ZAZ was supported by a Lundbeck Foundation Scholarship (The Danish Neurological Society) and a research grant from Rigshospitalets Forskningspuljer (E-23327-03). AC was supported by Foreningen til støtte af forskning ved Dansk Hovedpine Center. SLC was funded by the Candys Foundation and by BRIDGE – Translational Excellence Programme, Faculty of Health and Medical Sciences, University of Copenhagen, supported by the Novo Nordisk Foundation (Grant no. NNF23SA0087869).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: ZAZ, JT, LK, EG, ZH, MMK, AC, DMK, and JA have nothing to disclose. RDM has received personal fees from Novartis, Pfizer, and ADIUM Pharmaceuticals, outside of the submitted work. RDM is member of the Headache Study Group of the Mexican Academy of Neurology and serves as a Graphical Abstract Editor of Cephalalgia Reports. SLC has received personal fees from CEVA Animal Health, Deep Apple Therapeutics, Guidepoint, and Y-mAbs Therapeutics outside of the submitted work. MA has received personal fees from AbbVie, Amgen, Astra Zeneca, Eli Lilly, GlaxoSmithKline, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals, outside of the submitted work, and is currently Primary Investigator for ongoing clinical trials sponsored by AbbVie, Lundbeck and Pfizer. MA also serves as an Associate editor of Brain and The Journal of Headache and Pain.
Data availability statement
Upon reasonable request, the corresponding author can provide the necessary data and materials to interested researchers for the purpose of academic scrutiny, reproducibility, and further scientific investigation.
Supplemental material
Supplemental material for this article is available online.