
Abstract
Background
Second and third generation gepants have been recently approved for migraine therapy. They represent the first drugs that are able to work as both preventatives and symptomatics of the migraine attack. Their ability to counteract calcitonin gene-related peptide signaling has been convincingly shown, but where they act to exert the therapeutic effects remains unsolved. Although the low brain/plasma ratio suggests peripheral antimigraine activity of gepants, recent preclinical and clinical lines of evidence hint that these compounds may also act centrally.
Methods
By means of mass spectrometry analysis, we have measured the biodistribution of atogepant and rimegepant in plasma, dura mater, trigeminal ganglion (TG), parietal brain cortex and hypothalamus of mice. The biodistribution of oxazepam has been also determined as that of a prototypical brain permeant drug. Animals received interspecies (human-to-mouse) converted doses. Drugs were administered orally, as single or repeated (seven days) dosing. Atogepant was also administered as a single oral or intranasal dose matching (mg/kg) that adopted in migraine patients.
Results
Upon administration of interspecies converted oral doses, we found that atogepant reached similar Cmax in plasma and TG after three hours, that then rapidly decreased at six and 12 hours. Of note, atogepant contents in the parietal brain cortex linearly increased up to six hours (reaching a brain/plasma concentration ratio of 5.6) and substantially decreased at 12 hours. Tissue contents of rimegepant were lower than those of atogepant, although the drug reached in the brain Cmax analogues to those found in the TG. Three hours after dosing, we also found the highest accumulation of atogepant and rimegepant in the dura, with substantial accumulation even in the hypothalamus where drug contents equaled those present in the TG. Of note, when atogepant was administered orally or intranasally at a dose corresponding to that adopted in patients, it also reached brain contents comparable to those found in the TG. However, a preferred delivery of atogepant to the TG was obtained with the intranasal route. At variance with oxazepam, the two gepants did not accumulate in the TG or parietal brain cortex upon a seven day oral treatment.
Conclusions
The data obtained in the present study indicate substantial and transient permeation of the mouse brain by gepants.
This is a visual representation of the abstract.
Introduction
In spite of the high prevalence of migraine the neurobiology of the events concurring to its pathogenesis is still unresolved (1). A key question that needs to be answered is whether migraine pain originates at trigeminal afferent terminals within the neurovascular structures of the meninges (2) and/or it must be also related to a primary, functional derangement of central nervous system (CNS) regions involved in pain processing (3). If on the one hand several features of migraine pain suggest a peripheral origin, the widespread dysfunction of sensory processing that precedes migraine pain points to a primary involvement of the brain. In this scenario, a detailed understanding of the mechanisms of action of antimigraine drugs can provide important information about migraine pathogenesis.
The ability of anti-calcitonin gene-related peptide (CGRP) drugs (i.e. monoclonals and gepants) to prevent migraine occurrence, besides its remarkable clinical relevance, highlights the undisputed, causative role of CGRP in the genesis of the disorder of a substantial percentage of patients (4). As far as the anti-CGRP monoclonal antibodies are concerned, the difficulty of immunoglobulins to permeate the blood–brain barrier (BBB) has pointed to the meningeal trigeminovascular system as the monoclonals’ site of action (5,6). Still, the genuine migraine preventative effect of anti-CGRP monoclonal antibodies (7), their ability to prevent accompanying symptoms such as phono-photophobia, nausea-vomiting and aura (8,9), and their ability to ameliorate depressive symptoms independent from the anti-migraine effects (10,11) suggest that they might have central actions. As for gepants, they differentiate from analogous, target-specific, small molecules anti-migraine drugs such as triptans or ditans in light of their unique ability to function as both symptomatics and preventatives. Even for gepants, however, the site of action waits to be deciphered (12).
Gepants represent CGRP receptor antagonists recently approved for acute (ubrogepant, rimegepant and zavegepant) or preventative (rimegepant and atogepant) migraine treatment (13,14). Although gepants, as small molecules, should readily permeate the BBB, prior work tends to rule out a central mode of action. For example, a human positron emission tomography (PET) study reports that a therapeutically active dose of the first generation gepant telcagepant does not displace a structurally analog radiotracer from CGRP receptor occupancy in the brain (15). Also suggesting a peripheral site of action, preclinical findings indicate that systemic but not intracerebroventricular olcegepant counteracts cephalic allodynia in a mouse migraine model (16). This is in keeping with the ability of atogepant to reduce activation of meningeal nociceptors and their signaling to the trigeminal nucleus (17). Although these findings are of importance with respect to understanding how gepants counteract migraine pain, the question of how these drugs act to prevent migraine occurrence before the pain component develops remains to be clarified. In this respect, a very recent report shows the ability of ubrogepant to reduce prodromal symptoms such as photophobia, phonophobia, fatigue, neck pain, dizziness, difficulty concentrating and thinking in migraine patients (18). By definition, prodromal symptoms are CNS dysfunctions that anticipate migraine pain, suggesting that ubrogepant can target the brain, and in particular the hypothalamus, where prodromes are considered to take origin (19). These findings, however, are at odds with studies reporting limited penetration of atogepant (20) and ubrogepant (21) in the brain of rats or rhesus monkeys, respectively.
Notably, the CNS content of a drug per se is not informative of its possible central action, being the free, intersynaptic drug concentration and the inhibitory constant (Ki) at receptor targets the two parameters of relevance to infer neuromodulation. It is also worth noting that a comparison of the brain contents of gepants with those reached at peripheral tissues (i.e. trigeminal ganglion (TG) or dura) where they are supposed to act is lacking. On this basis, in the present study, we evaluated the contents of atogepant and rimegepant in different brain regions and peripheral structures or relevance to migraine pathogenesis in mice acutely or chronically exposed to oral dosing.
Methods
Animals
Adult male C57Bl/6J mice weighing 25–30 g (Charles River, Milan, Italy) were housed in groups with free access to food (Harlan Global Diet 2018; Harlan Laboratories, Udine, Italy) and water, and maintained under a 12:12 h light/dark photocycle at room temperature (21°C). All animal manipulations were performed according to the European Community guidelines for animal care (DL 116/92, Application of the European Communities Council Directive 86/609/EEC). Male mice were used to be consistent with prior work on evaluating biodistribution and effects of gepants in male mice (16), rats (17) and monkeys (15). Animals were randomized (generating groups by the RAND function of Excel (Microsoft Corp., Redmond, WA, USA)) and treated with atogepant (10.28 mg/kg) and rimegepant (12.84 mg/kg) by oral gavage adopting doses considering the classic human-to-mouse dose conversion factor of 12.3 (22). Atogepant was also administered as a single oral or intranasal dose of 0.85 mg/kg that exactly matches (in terms of mg/kg) that adopted in humans. The intranasal administration was performed by administering atogepant in a volume of 2 µl directly in the nostrils of mice sedated with 2,2,2-tribromethanol (250 mg/kg, i.p.) (23). A group of mice was also exposed to oxazepam (10.28 mg/kg) as our positive control. Each experiment was conducted with at least five mice per group given that prior study from our group detecting oxazepam in plasma, dura, TG, parietal brain cortex and hypothalamus of mice (24) proved this number sufficient to detect differences over time and among different drug treatments. Tissue drug contents and plasma concentrations have been determined either at one, three, six and 12 hours after a single dosing, or after a chronic exposure paradigm. During the chronic treatment, mice were daily dosed orally with atogepant (10.28 mg/kg), rimegepant (12.84 mg/kg) or oxazepam (10.28 mg/kg) and killed one hour or six hours after an additional dose on day 7.
At the given times after drug exposure, animals were killed with isoflurane, transcardially perfused (24) or not, and plasma and tissue specimens collected. Plasma was obtained from whole blood collected in an heparinized Eppendorf via the transcardial cannula before perfusion. Frontal and parietal dura mater were scraped from the calvarium, stretched and cut sagittally. Each hemidura was weighed and frozen for subsequent western blotting and drug content analysis. From each mouse, the right or left TG was randomly collected and divided into two sagittal pieces that were weighted and frozen for western blotting or drug content analysis. Brains were removed and snap frozen. Later on, they were cut with the cryostat to enable collection from coronal slices of the parietal cortex (randomly dissected from the right or left hemisphere) and hypothalamus. The brain regions of each mouse were collected from at least four slices (30 µm) at the level of the median eminence. Specimens of parietal cortex or hypothalamus dissected from sequential slices were processed for western blotting or mass spectrometry analysis.
Western blotting
Western blotting experiments were conducted as previously described (25) with minor modifications. Mice were anesthetized with zoletil/xylaxine and transcardially perfused or not with cold saline for 15 minutes as described previously (24). Specimens were collected in Eppendorf tubes as described above and dissolved in 1% sodium dodecyl sulfate (SDS). The BCA Protein Assay was used to quantify the total protein levels. Lysates (30 μg per lane of protein) were resolved by electrophoresis on a 4–20% SDS-polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA, USA) and transferred onto nitrocellulose membranes. After blocking with 5% milk, blots were incubated overnight at 4°C with a rabbit anti-hemoglobin alpha antibody (catalog. no. MA5-35794; Thermo Fisher Scientific, Milan, Italy) diluted 1:1000. Tubulin (PAS-29444; Invitrogen, Waltham, MA, USA) or GAPDH (G8795; Merck, Milan, Italy) were used as a loading control. Immunodetection was performed with horseradish peroxidase-conjugated secondary antibodies (diluted 1 : 2000) (Amersham Biosciences, Little Chalfont, UK) in Tris-buffered saline-Tween 20 containing 5% non-fat dry milk. After washing, the membranes were detected using chemiluminescence (ECL plus; Euroclone, Padova, Italy) visualized by Chemidoc (Bio-Rad Laboratories).
Liquid chromatography/mass spectrometry analysis
Plasma and tissue (trigeminal ganglion, parietal cerebral cortex, hypothalamus and dura madre) specimens were weighed, 200 µl of methanol (MeOH) was added and the samples were homogenized by sonication, and then centrifuged at 13,000 rpm × 17,949 g at 4 °C for 5 minutes. Supernatants were collected and stored at −20 °C. Rimegepant was used as the internal standard (IS) for the samples from atogepant-treated animals and, similarly, atogepant was used as the IS for the samples of rimegepant-treated animals. Two different, freshly prepared solutions of the two molecules (20 pg/µl) were prepared in 1 ml of liquid chromatography-mass spectrometry (LC-MS) grade water with 10 m
LC-MS/MS analyses were performed using a LC-tandem mass spectrometry system consisting of a HPLC series 200 micro pump apparatus, equipped with column oven and autosampler (PerkinElmer, Waltham, MA, USA), coupled to a 4000 Q Trap (MDS Sciex; Thornhill, ON, Canada) mass spectrometer equipped with a VSpray ESI interface, operating in multiple reaction monitoring mode (MRM) in positive ions. The LC column was a Zorbax 300 SB-C8 Narrow Bore (100 mm × 2.1 mm i.d., 3.5 μm particle size; Agilent Technologies, Santa Clara, CA, USA) with its column guard; eluents were 10 m
Chromatographic conditions.

List of chromatographic retention time (RT), selected multiple reaction monitoring mode parameters, declustering potential (DP), entrance potential (EP), collision energy (CE), cell exit potential (CXP) for each measured analyte.

A calibration curve for each compound was prepared using a blank matrix (plasma and brain) adding scalar amounts of the investigated molecule and the same volume of the IS used for samples to a final volume of 50 µl, as described for actual samples. The range for quantitative measurement was from 5 to 200 pg for atogepant and from 5 to 50 pg for rimegepant, expressed in total amount in the 5-µl injected volume. The peak area ratios of the quantifier ions (Table 2) of the investigated molecule was calculated; the obtained calibration curves were linear in the selected range of concentrations with r2 > 0.98 and 0.99 for atogepant and rimegepant, respectively. The concentration in the experimental samples was calculated considering the dilution of the original MeOH solution and, in the case of dura, trigeminal ganglion and brain, calculated for the weight of the anatomical part. The matrix effect was evaluated in six different blank matrices spiked with the two molecules and compared to a solution in water/MeOH 1:1 spiked with the same amount of the molecules: a suppression effect lower than 7% was observed in all the blank matrices; the stability of the IS peak areas was used as a control parameter to verify the stability of matrix effect in actual samples. The dwell time was 200 ms for all transitions.
Statistical analysis
Data are presented as the mean ± SEM. Differences among groups were performed using two-way analysis of variance followed by Tukey's test or Student's t-test. p < 0.05 was considered statistically significant. Statistical analyses were conducted using Prism, version 8 (GraphPad Software Inc., San Diego, CA, USA).
Results
Efficacy of blood removal from mouse tissues by transcardial perfusion
A potential confounding factor when parenchymal drug contents are evaluated is residual blood contamination after transcardial perfusion. To check for the efficacy of blood removal, we evaluated the presence of hemoglobin in perfused tissues. As previously reported (26), we found that, at variance with the dura, the parietal brain cortex and the hypothalamus, it was impossible to completely remove blood from the TG (Figure 1). We reason that the different perfusion efficiency between the TG and the other tissues can be ascribed to the reduced vascularization of the former compared to the latter.

Western blotting evaluation of the efficacy of blood removal by transcardial perfusion from peripheral and central structures of the mouse. The ability of transcardial perfusion to remove haemoglobin from dura mater (a), hypothalamus (b) TG and parietal brain cortex (c) is shown. Whole blood is shown as positive control. Blots are representative of at least six samples for each structure. GAPDH or tubulin are shown as a loading control. Ato = atogepant, Rim = rimegepant.
Biodistribution of atogepant and rimegepant after single or repeated dosing in mice
Atogepant, rimegepant and oxazepam contents have been measured at one, three, six and 12 hours after injection in the plasma, TG and parietal brain cortex. Drugs were delivered orally at a dose of 10.28 mg/kg (atogepant) and 12.84 mg/kg (rimegepant), equaling the human dose × 12 according to the human-mouse inter-species conversion factor (22). Oxazepam (also dosed orally at 10.28 mg/kg) was selected as a prototypical BBB penetrant drug as previously reported (24). Molecular structures of the two gepants and oxazepam, along with their index of lipophilicity (octanol/water partition coefficient, logP), are shown in Figure 2(a).
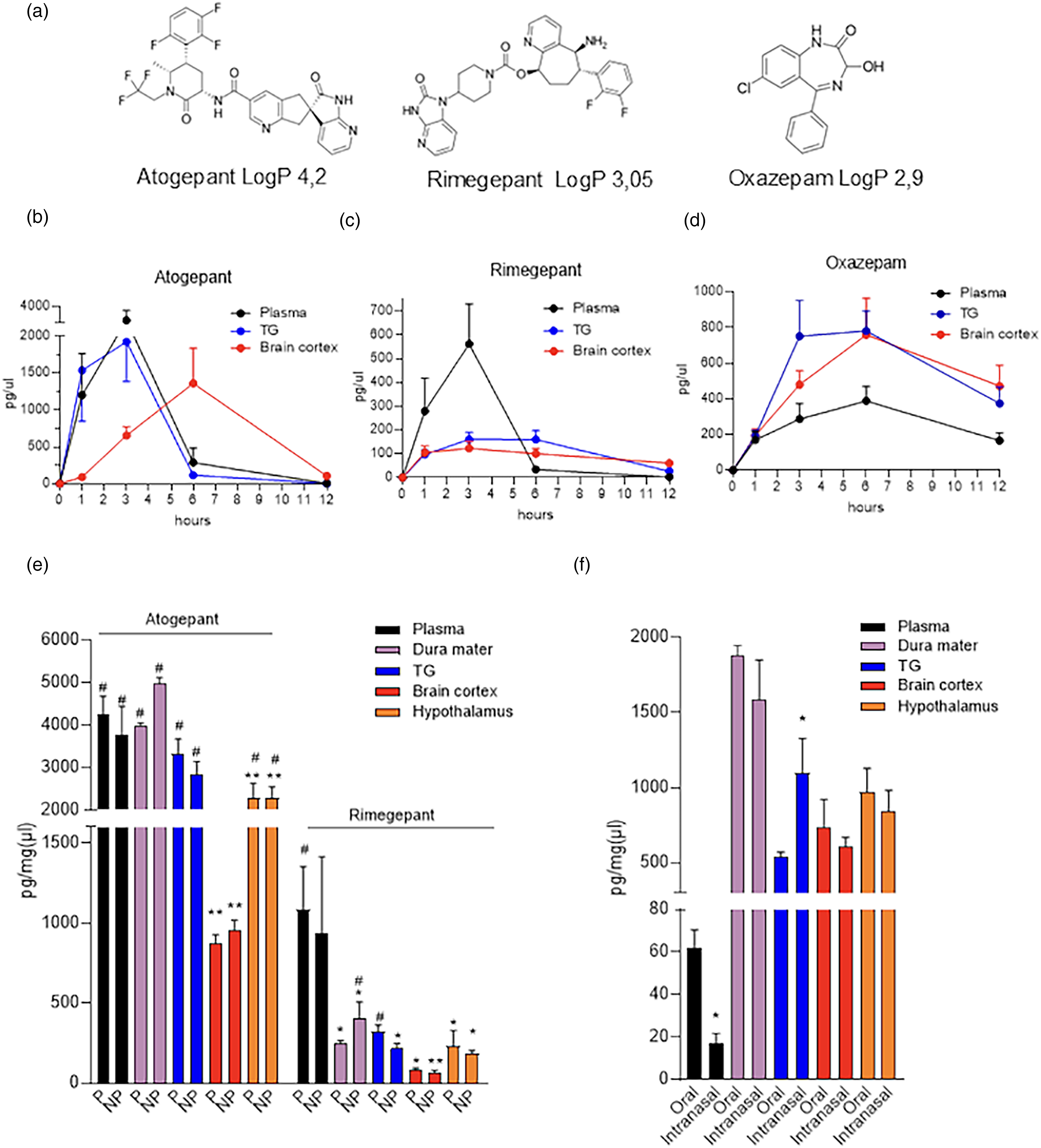
Biodistribution of atogepant and rimegepant in peripheral and central structures of mice exposed to single dosing. (a) Molecular structures of atogepant, rimegepant and oxazepam with their respective logP. Time course of the concentrations/contents of atogepant (b), rimegepant (c) and oxazepam (d) in plasma, TG and parietal brain cortex of mice treated with an oral dose of 10.28 mg/kg (atogepant and oxazepam) or 12.84 mg/kg (rimegepant). (e) Concentrations/contents of atogepant or rimegepant in transcardially perfused (P) or non-perfused (NP) dura mater, TG, parietal brain cortex and hypothalamus of mice three hours after an oral dose of 10.28 mg/kg (atogepant and oxazepam) or 12.84 mg/kg (rimegepant). (f) Concentrations/contents of atogepant in transcardially perfused dura mater, TG, parietal brain cortex and hypothalamus and the corresponding plasma of mice three hours after an oral or intranasal dose of 0.857 mg/kg. In (b) to (d), each point represents the mean ± SEM of at least five mice. In (e) and (f), each column represents the mean ± SEM of five (oral) or five (intranasal) mice. In (e), *p < 0.05, **p < 0.01 vs. plasma and #p < 0.05 vs. parietal brain cortex, two-way analysis of variance with Tukey's post-hoc test. In (f), *p < 0.05, ** p < 0.01 and *** p < 0.001 vs. oral, Student's t-test. TG = trigeminal ganglion.
We found that plasma contents of atogepant increased 1 h after dosing, reaching concentrations of 2742 ± 921 pg/µl at three hours. Drug contents rapidly decreased at six hours, and, at 12 hours, they were almost undetectable (Figure 2(b)). Interestingly, atogepant contents in the TG showed temporal kinetics and values similar to those found in plasma (Figure 2(b)). By contrast, cortical contents of atogepant were very low one hour after dosing, but then linearly increased up to six hours reaching contents of 1361 ± 422 pg/mg (Figure 2(b)). Surprisingly, we found that rimegepant had different biodistribution compared to atogepant. In particular, in spite of the very similar doses of rimegepant and atogepant administered to mice (i.e. 12.84 vs. 10.28 mg/kg, respectively), animals receiving rimegepant showed a much lower increase in plasma concentrations, with a peak of 562 ± 164 pg/µl three hours after dosing (Figure 2(c)). At variance with atogepant, the TG contents of rimegepant did not parallel the plasma concentrations, showing a flattened but more prolonged increase with a concentration peak of 160 ± 18 pg/mg at three hours. Rimegepant distribution in the parietal brain cortex had a temporal pattern similar to that of the TG, with a peak of 123 ± 19 pg/mg at three hours (Figure 2(c)). As for oxazepam, we found that the plasma concentrations increased at one and three hours, peaked at six hours with values of 388 ± 49 pg/µl, and appeared almost halved (166 ± 31 pg/mg) at 12 hours (Figure 2(d)). Oxazepam contents in the TG and parietal brain cortex were higher than those of plasma and peaked to 782 ± 104 and 759 ± 132 pg/mg after six hours, respectively (Figure 2(d)). After an additional six hours, the contents of oxazepam in TG and parietal brain cortex were halved (Figure 2(d)), in keeping with the drug t1/2 of five to seven hours in mice (27).
Data showing that the contents of gepants in the TG had a low degree of variability in spite of the considerable variations in residual hemoglobin suggested that blood contamination does not significantly affect tissue drug contents. To address this issue, we compared atogepant and rimegepant contents in the dura mater, TG, parietal brain cortex and the hypothalamus of mice exposed to atogepant or rimegepant for three hours and then undergoing (or not) transcardial perfusion. Figure 2(e) shows that atogepant reached in the dura contents (pg/mg) analogues to those of plasma (pg/µl). Confirming a reduced biodistribution of rimegepant compared to atogepant in the mouse, the dura of animals receiving rimegepant showed drug contents lower that the corresponding concentrations of plasma (Figure 2(e)). Surprisingly, however, we found that TG, dura, cortical and hypothalamic drug contents did not differ in perfused and not perfused animals receiving atogepant or rimegepant. Remarkably, parietal brain cortex contents of atogepant were lower than those of other tissues including plasma, whereas a tendency was found for rimegepant (Figure 2(e)).
The considerable contents reached by atogepant in the different tissues prompted us to evaluate its biodistribution adopting oral doses of 0.857 mg/kg, matching those administered to patients (60 mg/70 kg). Furthermore, in light of the possible, favored TG delivery of gepants dosed intranasally (28), we also evaluated the biodistribution of atogepant administered at 0.857 mg/kg in a volume of 2 µl directly in the nostrils of mice. Interestingly, we found that three hours after the administration and transcardial perfusion, the contents of atogepant in the dura, parietal brain cortex and hypothalamus were similar in mice receiving the drug orally or intranasally (Figure 2(f)). Conversely, animals receiving intranasal administration showed lower plasma concentrations but higher TG contents (Figure 2(f)). Again, the highest drug contents were found in the dura, with the TG, parietal brain cortex and hypothalamus displaying similar values (Figure 2(f)).
In light of the use of atogepant and rimegepant in chronic treatment schedules for migraine prevention (13,14), we also attempted to understand whether their repetitive administration at interspecies converted doses leads to tissue drug accumulation. To this end, drug contents were measured in plasma, TG and parietal brain cortex of mice exposed to daily atogepant, rimegepant or oxazepam for six days and killed the following day, one or six hours after an additional dose (therefore defined as ‘6 days+1h’ and ‘6 days+6h’). Interestingly, we found that chronic exposure to gepants minimally affected their tissue drug contents and biodistribution. Specifically, plasma concentrations and tissue contents of atogepant or rimegepant did not differ between ‘6 days+1h’ and ‘1h’ mice (Figure 3(a)). Conversely, a robust increase in oxazepam contents was detected in plasma, TG and parietal brain cortex of ‘6 days+1h’ compared to ‘1h’ mice (Figure 3(a)). Similar findings were obtained when contents of atogepant and rimegepant were compared in ‘6 days+6h’ and ‘6h’ mice, with the exception of the rimegepant brain contents that were higher in mice undergone chronic exposure (Figure 3(b)). As expected, the contents of oxazepam further increased in the plasma, TG and parietal brain cortex of mice exposed to the ‘6 days+6h’ treatment schedule (Figure 3(b)).

Biodistribution of atogepant and rimegepant in peripheral and central structures of mice exposed to repeated dosing. Concentrations/contents of atogepant, rimegepant or oxazepam in plasma, TG and parietal brain cortex of transcardially perfused mice exposed to a six day treatment with oral doses of 10.28 mg/kg (atogepant and oxazepam) or 12.84 mg/kg (rimegepant). In (a), plasma concentrations or tissue drug contents of mice exposed to a six day treatment and killed 1 h after an additional dose on day 7 (6 day+1 h) are compared with those present in mice exposed to a single treatment of one hour. In (b), plasma concentrations or tissue drug contents of mice exposed to a six day treatment and sacrificed six hours after an additional dose on day 7 (6 day+6 h) are compared to those present in mice exposed to a single treatment of six hours. In (a) and (b), each column represents the mean of at least five mice. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. one hour (a) or six hours (b), Student's t-test. TG = trigeminal ganglion.
Discussion
To the best of our knowledge, the present study is the first that compares bioavailability of atogepant and rimegepant in the dura, TG, parietal brain cortex and hypothalamus of mice. By doing so, it provides key information of the relative contents that gepants reach in peripheral and central nervous system structures involved in the pathogenesis of migraine.
Although a convincing understanding of the mechanism of action of gepants is currently lacking, they are reported to be unable to permeate the BBB (29). Admittedly, clear cut, scientific arguments in support of this pharmacokinetic barrier still need to be provided. One of the claim of the difficulty of these drugs to reach the brain parenchyma is based on their theoretical, unfavorable partition coefficient (logP, 4.2 for atogepant and 3.5 for rimegepant) (29). These values define high degrees of lipophilicity that in principle help to permeate the BBB (30). They are also well in the range of those of rapidly acting neuroactive drugs such as fentanyl and midazolam, endowed with logP values of 4.05 (31) and 3.6 (32), respectively. The low brain/plasma concentration ratio of gepants has been also considered an indication of their poor BBB permeability. For example, Moore et al. (20) report brain/plasma concentration ratios of atogepant of 0.013 and 0.020 in rats exposed to a single oral dose of 5 or 20 mg/kg, respectively. They conclude, therefore, that brain penetrance of atogepant is extremely limited. This assumption is in contrast with the present study conducted in mice challenged with a similar oral dose of atogepant (10.28 mg/kg). Indeed, although, at one hour post dosing, we also detected a low brain/plasma concentration ratio, we found that atogepant accumulates in the brain at later time points, while rapidly disappearing from the plasma. Specifically, six hours after dosing the brain/plasma concentration ratio of atogepant increases to 5.6. The low brain/plasma concentration ratio reported by Moore et al. (20) is calculated at the plasma Cmax. Unfortunately, Moore et al. (20) report neither the time point corresponding to Cmax, nor the sensitivity of their analytical method. Hence, a single time point might be insufficient to address the BBB permeability of a drug that accumulates in the brain over time. It is also worth noting that claiming that a drug does not enter the brain because of a low brain/plasma concentration ratio may be misleading if the concentration ratio between plasma and peripheral tissues is not determined. In this regard, we report here that the TG/plasma and brain/plasma concentration ratios are in the same range for both atogepant and rimegepant. However, the rapid accumulation of atogepant in the TG compared to the slower increase in the brain is a clear index of the partial impediment for the drug to cross the BBB. It is also worth noting that, in keeping with the rapid drug metabolism in rodents, atogepant shows a plasma t1/2 of approximately four hours that corresponds to one-third of that reported in humans (11 hours) (33). In this regard, we reason that a higher ability to metabolize rimegepant than atogepant, as well as a more pronounced absorption/stability of atogepant, could explain the lower bioavailability of rimegepant in mice. Indeed, although atogepant and rimegepant have similar moieties and a t1/2 of approximately 11 hours in humans (29), the trifluoromethyl group of atogepant confers high lipid solubility, metabolic stability and bioavailability (34). Also, the amine group exclusively present in rimegepant and added to improve water solubility reduces per se bioavailability (35). The lower bioavailability of rimegepant in rodents (45%) (35) than in humans (67%) (36) is also in keeping with our findings. As for oxazepam, we found delayed absorption compared to both gepants, as well as time-dependent brain accumulation in keeping with its central effects. A striking difference between gepants and oxazepam, however, emerged when we evaluated the tissue drug contents in mice undergoing a chronic exposure paradigm. Indeed, at variance with oxazepam, a six day exposure to atogepant or rimegepant did not prompt tissue drug accumulation (with the exception of cortical rimegepant). These findings indicate the ability of mice to rapidly excrete gepants and are in keeping with evidence that these drugs do not accumulate upon repetitive administration in humans (13,29).
Somehow unexpectedly, we report that lack of transcardial perfusion did not alter gepant contents in the TG, dura mater, parietal brain cortex and hypothalamus. Importantly, we also found that hypothalamic contents of atogepant and rimegepant were almost two-fold greater than those of the parietal brain cortex, and analogues to those of TG. The leaky BBB of the hypothalamus (37) is in keeping with this finding. As for the irrelevant role of brain perfusion, we emphasize that the brain has a very low vascular space that in the mouse equals approximately 10 μl/g, or 1/100 of tissue if we adopt a weight to volume conversion (38). It is therefore conceivable that the plasma concentrations of atogepant and rimegepant divided by a factor of 100 do not affect drug contents of the brain parenchyma (note that values are expressed per mg of proteins). The same reasoning might be carried out for the TG that should be even less vascularized than the brain. Of note, irrelevance of brain perfusion corroborates penetrance of gepants within the brain parenchyma. This assumption is further corroborated by the findings that atogepant, when administered either orally or intranasally at doses disregarding the interspecies conversion factor and identical to the human ones (in terms of mg/kg), also reaches brain concentrations similar to those found in the TG. Of note, these findings support the hypothesis of a facilitated TG delivery of intranasal gepants (28), also indicating their ability to readily permeate the brain when administered via the nasal route.
The ability of gepants to reach the brain parenchyma is in keeping with that of olcegepant to modulate activity of second and third trigeminovascular neurons (39,40), as well as brainstem structures involved in descending inhibition of pain such as the periaqueductal gray matter (41). Even detection of ubrogepant in the human cerebrospinal fluid (42) suggests brain penetrance of gepants. Possibly more indicative of central effects of gepants is a very recent report showing that ubrogepant counteracts migraine prodromal symptoms such as photo-phonophobia, dizziness, and difficulty thinking and concentrating when taken by patients before headache initiation (18). Adopting a more general and clinical perspective, if the migraine attack originates within the brain, then the preventative efficacy of gepants would be per se proof of central actions. In this regard, however, prior work is at odds with evidence that targeting of the brain by gepants is clinically relevant. For example, a PET study performed in humans by Hostetler et al. (15) reports that telcagepant does not displace the radioligand and CGRPR tracer [11C]MK-4232 when administered at doses known to counteract migraine, being able to prompt radioligand displacement only at supratherapeutic doses. In this study, however, there is no proof that signal from the radioligand comes from CGRPR binding (i.e. is receptor specific). Indeed, [11C]MK-4232 also shows high affinity for the human amylin receptor (15). Although CGRP-dependent displacement of [11C]MK-4232 in humans is impracticable, it could have been performed in the in vitro experiments on the radioligand binding in monkey brain slices (15). Adopting an opposite perspective, the structural homology between telcagepant and [11C]MK-4232, along with the very rapid brain uptake of the latter (15), corroborates permeation of the human BBB by gepants. Additional work suggests that olcegepant counteracts cephalic allodynia in a migraine model in mice when administered systemically but not intracerebroventricularly (16), again suggesting peripherally-restricted pharmacodynamics actions. However, any claim that these findings indicate a lack of central actions may be premature given that how olcegepant diffuses into the brain parenchyma from the ependyma in comparison to brain capillaries is unknown.
Conclusions
In conclusion, the present study provides evidence that gepants reach the mouse brain when administered orally or intranasally. Additional studies are needed to define gepant contents in brain regions in addition to the parietal brain cortex and hypothalamus. In light of the emerging lower efficacy of gepants in male than female patients (43,44), it will be worth investigating whether gender affects brain penetrance of this class of drugs. Thus, future studies should be designed to incorporate both sexes aiming to understand whether gepant pharmacokinetics differs between males and females. Whether a gepant central effect is of therapeutic relevance and synergizes with their effects at meningeal trigeminovascular terminals and TG neurons (17) remains to be determined. Should gepants exert antimigraine effects also through a central action, it would be of paramount importance determining the neuronal populations whose pharmacological targeting prevents and aborts migraine.
Article highlights
Atogepant and Rimegepant transiently accumulate in the mouse brain reaching concentrations similar to those found in the TG.
This occurs when animals are challenged with oral or intranasal doses either considering the human-to-mouse interspecies conversion or perfectly matching the human dose.
Data suggest analogous biodistribution of gepants in the TG and brain of mice.
Footnotes
Author contributions
All authors made substantial contributions to conception and study design. Alessandra Pistolesi and Daniela Buonvicino performed experiments and collected data. Francesco De Cesaris, Daniela Buonvicino and Alberto Chiarugi designed the experiments and discussed data. Daniela Buonvicino and Alberto Chiarugi wrote the manuscript.
Data availability statement
Data and materials are available upon request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical statement
All animal manipulations were performed according to the European Community guidelines for animal care.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The study was supported by grants from Agenzia Italiana Ricerca contro il Cancro (AIRC), Italian Foundation for Multiple Sclerosis (FISM, 2022/R-single/023) and The PNRR project (CN3-Gene therapy and RNA technology Drugs; THE - Health Ecosystem).