
Abstract
Background: The ATP-sensitive K+ (KATP) channel openers levcromakalim and pinacidil are vasodilators that induce headache in healthy people. The neuropeptide calcitonin gene-related peptide (CGRP) induces headache in healthy people and migraine in migraineurs, potentially through a mechanism that involves opening of vascular or neuronal KATP channels and mast cell degranulation. Using rat as a model, we studied the molecular presence of KATP channels in the trigeminovascular system. Furthermore, we examined whether KATP channel openers stimulate the in vitro release of CGRP and whether they degranulate dural mast cells.
Methods: mRNA and protein expression of KATP channel subunits were studied in the trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC) by qPCR and western blotting. In vitro CGRP release was studied after application of levcromakalim (1 µM) and diazoxide (10 µM) to freshly isolated rat dura mater, TG and TNC. Rat dural mast cells were challenged in situ with levcromakalim (10−5 M) to study its potential degranulation effect.
Results: mRNA and protein of KATP channel subunits Kir6.1, Kir6.2, SUR1 and SUR2B were identified in the TG and TNC. KATP channel openers did not release or inhibit capsaicin-induced CGRP release from dura mater, TG or TNC. They did also not induce dural mast cell degranulation.
Conclusions: KATP channel openers do not interact with CGRP release or mast cell degranulation. Activation of these channels in the CNS is antinociceptive and therefore cannot explain the headache induced by KATP channel openers. Thus, they are likely to induce headache by interaction with extracerebral KATP channels, probably the SUR2B isoforms.
Keywords
Introduction
The KATP channel openers levcromakalim and pinacidil induce headache as a side effect in clinical studies of asthma and hypertension (1–5). According to our previous experience with other substances, KATP channel openers would probably cause migraine attacks in migraine sufferers, although this has not been directly studied. The sensory signalling peptide calcitonin-gene related peptide (CGRP) triggers migraine after intravenous infusion, and CGRP receptor antagonists are effective in the acute treatment of migraine. Furthermore, KATP channel blockers attenuate the effect of CGRP on dural arteries (6).
KATP channel expression has so far not been studied in the trigeminal ganglion (TG) or the trigeminal nucleus caudalis (TNC), two key structures in migraine pain. Furthermore, it needs to be determined whether KATP channel modulation has an effect on CGRP release in the trigeminovascular system and on mast cell degranulation, another putative mechanism of migraine. To address these issues, we studied the mRNA and protein expression of KATP channel subunits in the rat trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC).
We have previously suggested that KATP channel openers cause headache via a vasodilatory effect on cerebral and dural arteries (7,8). In the present paper our aim was to exclude an action of the classical KATP channel openers levcromakalim and diazoxide on CGRP release from rat dura mater, TG and TNC and on degranulation of rat dural and peritoneal mast cells.
Materials and methods
All experimental procedures were performed in accordance with domestic guidelines and regulations for animal care and treatment. All adult male Sprague-Dawley rats were maintained in cages with a 12-h light/dark cycle with free access to food and water. The study protocol was approved by The Danish Animal Experimentation Inspectorate (file: 2004-561-850 and 2009-561-1664).
mRNA expression studies
Preparation of tissue RNA
A total of nine male Sprague-Dawley rats (320–350 g, Taconic M&B, Denmark) were anaesthetized with pentobarbital (Mebumal 60 mg kg−1), and the hindpaw withdrawal reflex was used to evaluate the depth of anaesthesia. Once anaesthetized, the rats were perfused transcardially with an ice-cold buffer solution ((mM): NaCl 119, NaHCO3 15, KCl 4.6, CaCl2 1.5, NaH2PO4 1.2, MgCl2 1.2, and glucose 5.5) to eliminate any blood in sample preparations. TGs and TNCs were dissected and placed in sterile Eppendorf tubes containing a RNA stabilization solution (RNAlater RNA Stabilization Reagent (Qiagen)). Three batches were prepared for analysis, each batch consisting of tissues collected and pooled from a group of three rats. All collected samples were kept in RNAlater overnight at 4°C and either further processed or stored at – 80°C for later use. RNA was purified from all the RNA stabilized samples using a RNeasy Lipid Kit (Qiagen), and subsequently the quantity and quality of the eluted RNAs were assessed on a NanoDrop spectrophotometer.
Reverse transcription-polymerase chain reaction (RT-PCR)
cDNA was synthesized from all purified RNA samples using the QuantiTect Reverse Transcription Kit and protocol from Qiagen, and all samples were subjected to the exact same RT-PCR mixture and thermal cycling conditions.
Quantitative real-time PCR (qPCR)
qPCR was performed with LightCycler technology (Roche) and SYBR Green I dye chemistry (SYBR Green JumpStart Taq ReadyMix (Sigma)), using our previous qPCR protocol and KATP channel subunit specific qPCR primers (7).
Experimental design and data analysis
mRNA was quantified using a calibrator-normalized relative quantification approach with efficiency correction. As previously, actin was used as a non-regulated reference gene for all relative expression calculations and a calibrator sample was prepared by collecting and mixing small chunks of heart from several rats and processing these for qPCR analysis as described above. Thus heart cDNA served as a calibrator sample in all studies and for final results, the expression levels of each KATP channel subunit in all the dissected tissues were calculated relative to their expression levels within the rat heart.
Protein expression studies
Western blotting
TGs and TNCs were isolated from three Sprague-Dawley rats, and the western blotting procedure was carried out exactly as described previously (8). In short, proteins were extracted from all tissues and separated by SDS polyacrylamide gel electrophoresis. After blotting the proteins onto a polyvinylidene difluoride membrane, the bound proteins were exposed to KATP channel subunit-specific antibodies.
In vitro CGRP release from the TG and the TNC
Preparation of the TGs and TNCs
For studies of CGRP release, rats were killed by CO2 inhalation and decapitated. The head was separated from the body, the skin was removed and the skull split along the sagittal plane. The brain was removed while the cranial dura mater was left attached to the two skull halves. The trigeminal ganglia were harvested by dissection 1 mm proximal and distal to the point at which the mandibular nerve branches off. The dura mater was carefully removed from the ganglia. For preparation of the TNC the head was separated from the body at the atlanto-occipital joint, the skin was removed and the skull split along the sagittal plane. The brain along with the brain stem was removed. The TNC running caudally from approximately 13–16 mm from the bregma was then isolated. The two TGs and the two TNCs were transferred to a beaker, and continuously washed for a minimum of 30 min with 500 ml synthetic interstitial fluid (SIF) of the following composition (in mM): 108 NaCl, 3.48 KCl, 3.5 MgSO4, 26 NaHCO3, 11.7 NaH2PO4, 1.5 CaCl2, 9.6 Na+ gluconate, 5.55 glucose and 7.6 sucrose.
In vitro effect of KATP channel drugs on CGRP release
Each TG and TNC was placed in 250 μl SIF in a cap of Eppendorf reaction tubes at 37°C. The specimens were washed with 250 μl SIF every 10 min (using a micropipette) while being careful not to touch the tissue. As a standard protocol, 200 μl of the sample was collected as a control after the fifth wash. Although there was some variation in control CGRP levels obtained from different rats, the CGRP levels detected in the two TGs or the two TNCs obtained from the same animal were very similar. Therefore it was possible to use one TG or one TNC as a control from each rat, thus reducing the experimental variations. Levcromakalim and diazoxide (1 and 10 µM, respectively) were added to the isolated tissues. SIF was collected after a 10 min incubation period for CGRP analysis. At the end of each experiment the maximum CGRP release was obtained by a challenge with 0.1 μM capsaicin. When capsaicin-induced CGRP release was studied in the presence of levcromakalim or diazoxide, the KATP channel openers were applied to the tissues 10 min before and during the capsaicin challenge.
Measurement of CGRP release
For measurement of the immunoreactive CGRP (iCGRP) content in the samples, an enzyme-linked immunoassay (EIA) kit (SPIbio, Paris, France) was used. To prevent CGRP degradation the samples were immediately transferred to a vial containing EIA-buffer with peptidase inhibitors. Samples were stored at −20°C and analysed within 1 week. The antibody in the iCGRP-EIA kit is directed against human-CGRP α/β but has 100% cross-reactivity to rat and mouse CGRP. The iCGRP detection level is about 2 pg/ml. Procedures were done in accordance to the kit manual. In short, the samples were incubated in wells at 4°C for 16–20 h. After the incubation, the wells were washed again and incubated with Ellman's reagent, an indicator. The wells were covered with aluminium foil and placed in a dark room for 60 min at room temperature. The optical density was measured at 410 nm using a microplate photometer (Tecan, infinite M200, software SW Magellan v.6.3).
Data analysis
Data analysis for the release of CGRP (in pg/ml) from the isolated dura mater, TG and TNCs (measured as immunoreactive CGRP in the samples) is given as mean values ± SEM. Absorbance was recorded and values were calculated through an interpolation method using an equation derived from the standard curve. Wilcoxon matched pairs test was used for nonparametric analysis of paired data. Differences were considered significant if p < 0.05. GraphPrism (GraphPad Software Inc., San Diego, CA, USA) was used for statistical analysis.
Drugs
All KATP channel drugs were purchased from Tocris Cookson Inc., UK. KATP channel openers levcromakalim and diazoxide were dissolved in dimethyl sulphoxide (DMSO) to provide stock solutions of 10 mM. Working concentrations of drugs were prepared by diluting the stock solutions in SIF. Capsaicin (Sigma Aldrich, Schnelldorf, Germany) was dissolved in 90% ethanol to obtain 10 mM and further dilutions were prepared in SIF.
Mast cell degranulation studies
Dural mast cell degranulation assay
Male Sprague-Dawley rats (300–400 g) were exsanguinated under CO2 anaesthesia (9). The preparations were prepared as described earlier (10). Briefly, after decapitation, skin, jaw and cranial muscles were removed from the head. The exposed skull was divided in the midline sagittal plane using a metal saw. Brain halves were carefully removed without disturbing the dura mater. The skull halves were transferred to a beaker of buffer (137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 0.5 mM CaCl2, 0.4 mM NaH2PO4, 5.6 mM glucose and 10 mM HEPES), and superfused with continuous flow of 500 ml buffer over at least 30 min.
The skull halves were transferred to concave moulds of clay lined with Vaseline to prevent leakage, 350 µl buffer was added and they were placed in a humid heating cabinet at 37°C. The buffer was changed 6 times at 5 min interval before starting the experiment.
To establish the baseline for each preparation, skull halves were incubated 2 × 15 min with normal buffer (350 µl), saving all aliquots of buffer on ice in Eppendorf tubes for analysis. Preparations were incubated with 350 µl vehicle or test substance dissolved in buffer for 2 × 15 min. Drug concentrations of 10−5 M were used for all tests.
Samples were centrifuged at 1000 g, 4°C for 5 min to sediment any erythrocytes and other cells. 50 µl of each supernatant in duplex was mixed with 60 µl 0.08 M citric acid buffer, pH 4.5 containing 8 mM 4-nitrophenyl-N-acetyl-β-
Peritoneal mast cell degranulation assay
Male Sprague-Dawley rats (300–400 g) were exsanguinated under CO2 anaesthesia. 20 ml oxygenated buffer (137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 0.5 mM CaCl2, 0.4 mM NaH2PO4, 5.6 mM glucose and 10 mM HEPES) was injected in the peritoneum, which was massaged gently for 3 min. The peritoneum was opened by midline section, and lavage was removed using a pipette. Cells were sedimented by centrifugation for 5 min at 400 g at 13°C. Sedimented cells were resuspended in 10 ml of the same buffer, washed and sedimented by centrifugation three times.
A bovine serum albumin (BSA)-Percoll density gradient was mixed in a Falcon tube (162 µl 35% BSA (Sigma Aldrich, St Louis, MO, USA), 8.10 ml Percoll® (GE Healthcare, Little Chalfont, Buckinghamshire, UK), 580 µl distilled water plus 1.16 ml salt solution (1.54 M NaCl, 27 mM KCl, 3.8 mM CaCl2). The cell pellet was resuspended in 5 ml physiological buffer and layered on top of the Percoll. Cells were separated by centrifugation at 225 G for 25 min at 13°C. The density gradient was discarded, and the pellet was washed three times in the buffer previously used. The pellet was finally resuspended in 1 ml buffer, 25 µl was stained with 5 µl 0.25% toluidine blue at pH 2 and the number of mast cells was counted under microscope. The cell suspension was diluted to 10 ml with buffer again, sedimented by centrifugation and diluted with RPMI1640 medium without phenol red (Labinova AV, Upplands Väsby, Sweden) to achieve a mast cell concentration of 400.000 mast cells per ml (20,000 mast cells per 50 µl). 50 µl aliquots of cell suspension in media were distributed in a 96-well plate (Nunclon™ Δ Surface, Nunc, Denmark). Three wells contained only 50 µl vehicle (blank standard samples) and 3 wells contained 50 µl media + 20 µl 1.2% Triton X 100 (Sigma-Aldrich, St Louis, MO, USA) (maximum degranulation samples). The remaining wells contained 50 µl of test-substance dissolved in RPMI1640, tests in duplicate in logarithmically increasing concentration steps. The plate was incubated for 30 min at 37°C and centrifuged at 1000 G for 5 min at 4°C to stop the reaction. 50 µl of the supernatant of each well (60 µl of the wells with Triton-X) was transferred to a new plate, and the wells without Triton-X were added 10 µl 1.2% Triton-X. To each well 60 µl 0.08 M citric acid buffer, pH 4.5 containing 8 mM 4-nitrophenyl-N-acetyl-β-
Data analysis
Dural mast cell degranulation assay
Degranulation was calculated relatively to the measured baseline release. The baseline was established as the average for the two performed baseline readings. Because of natural depletion, this preparation releases lower amounts of mast cell contents for each wash, which is why the control group gives a significantly lower degranulation than the baseline reading. Values are given as means ± SEM.
Peritoneal mast cell degranulation assay
Degranulation was calculated as degranulation of sample (Aassay well) in percentage of maximum degranulation (Alysed well) using the following formula ((Aassay well - Ablank)/(Alysed well – Ablank)) × 100%. Values are given as means ± SEM.
Drugs
Levcromakalim and diazoxide (Tocris Cookson Inc., UK) were dissolved in dimethyl sulphoxide (DMSO) to provide stock solutions of 10 mM. Working concentrations of drugs were prepared by diluting the stock solutions in SIF. Compound (48/80) (Sigma-Aldrich) a polymer produced by the condensation of N-methyl-p-methoxyphenethylamine with formaldehyde, was dissolved to 10−2 M in distilled water and further diluted in the buffer solution.
Results
mRNA expression studies in rat TG and TNC
mRNA transcripts of SUR1, SUR2B, Kir6.1 and Kir6.2 were detected in both rat TG and TNC, and expressions tended to be higher in the TNC (Figure 1). SUR2A mRNA was hardly detected in any of the tissues.
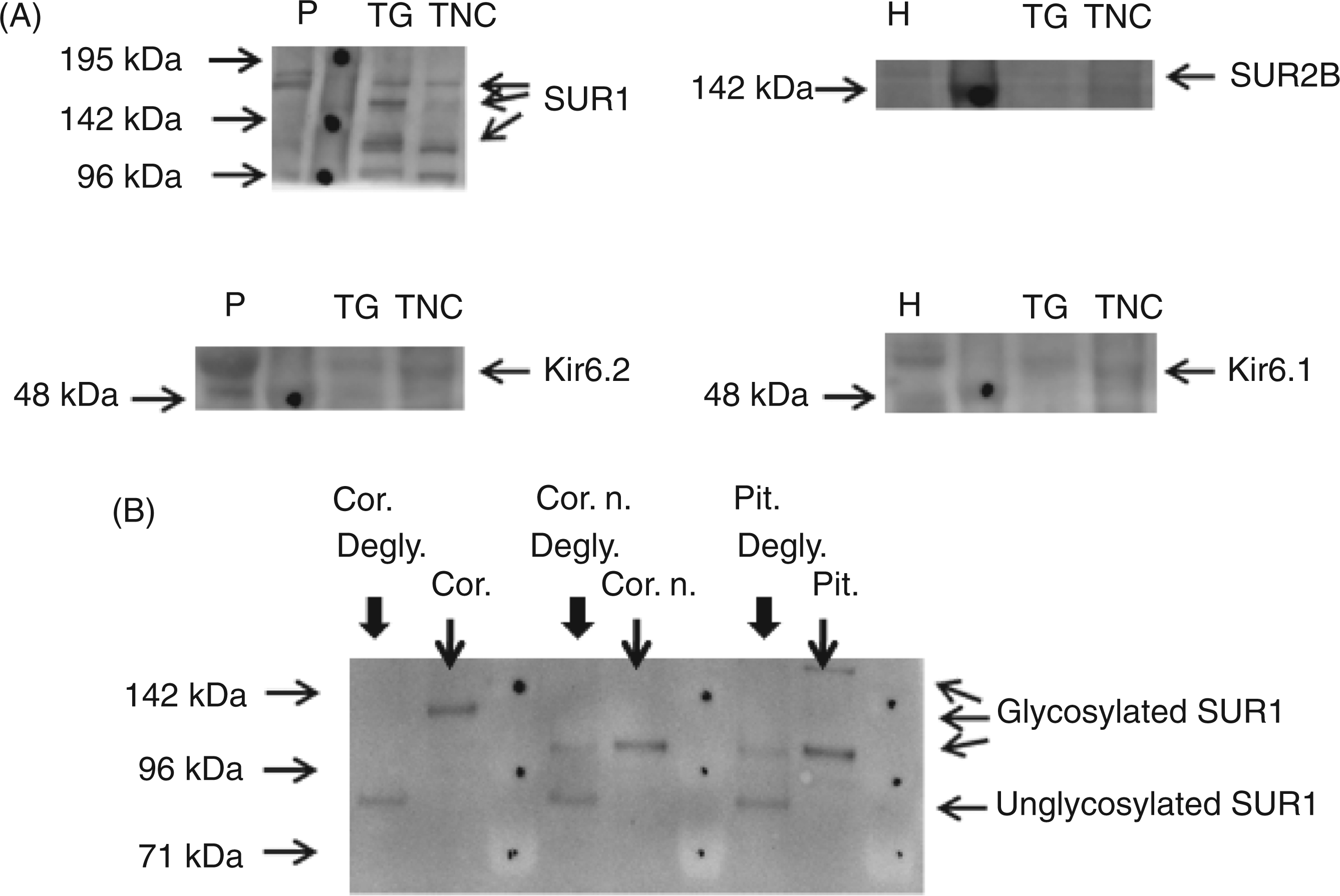
Relative mRNA expression of KATP channel subunits (A) SUR1, (B) SUR2A, (C) SUR2B, (D) Kir6.1 and (E) Kir6.2 in rat TG and TNC as determined by qPCR. For all tissues, n = 3 (each n represents TG and TNC chunks collected, mixed and processed for qPCR from a group of three rats). Expression levels are normalized ratios, e.g. the expression level of each KATP channel subunit in TG and TNC is related to their expression level in a heart calibrator sample (heart calibrator value = 10,000, marked in each figure). See Materials and methods for further details. Data expressed as mean values ± SEM. (A) Western blots showing the protein expression of KATP channel subunits SUR1, SUR2B, Kir6.2 and Kir6.1 in rat TG and TNC. Results from one out of three rats are shown here, but all were identical. Sizes of protein markers, shown as dots on the images, are marked to the left of each blot. Positive control lysates from pancreas (P) and heart (H) were loaded in lane 1 in order to identify each subunit. Detected subunit bands are shown with arrows to the right of each blot. Multiple SUR1 bands were detected in both tissues. (B) Western blot showing that SUR1 protein bands of different sizes are detected by the SUR1 antibody dependent on its tissue-specific glycosylation pattern. Protein lysates (15 µg) from mouse cortex (cor.), primary cultures of mouse cortical neurons (cor. n.) and mouse pituitary gland (pit.) were analysed in lane 2, 4, and 6, respectively. After enzymatic deglycosylation (degly., lane 1, 3 and 5, respectively) all SUR1 bands were pulled down and identified around 90 kDa, corresponding to unglycosylated immature SUR1. In vitro release of CGRP in rat dura mater. (A) Levcromakalim (1 µM) failed to release CGRP above basal levels; (B) Levcromakalim (LK) (1 µM) failed to inhibit CGRP release induced by capsaicin (0.1 µM) (Cap). Values are given as mean ± SEM. The Wilcoxon matched paired test was used for nonparametric analysis of paired data; *p < 0.05 as compared with basal CGRP release (n = 6).

Protein expression studies in rat TG and TNC
SUR1, SUR2B, Kir6.1 and Kir6.2 protein was detected in both rat TG and TNC, although SUR2B protein bands appeared faint. SUR1 blots revealed several SUR1 bands in both tissues, corresponding to differentially glycosylated SUR1 proteins (Figure 2A). Deglycosylation experiments performed on rodent tissues verified that the tissue-specific glycosylation of SUR1 appears on western blots as a spread of multiple bands of different sizes that can be shifted to similar sized bands after deglycosylation (Figure 2B).
In vitro CGRP release studies
In vitro release of CGRP from rat TG. (A) Levcromakalim (1 µM) and (B) diazoxide (10 µM) failed to release CGRP above basal levels. (C) Levcromakalim (LK) (1 µM) and (D) diazoxide (10 µM) (D) failed to inhibit CGRP release induced by capsaicin (0.1 µM) (Cap). Values are given as mean ± SEM. The Wilcoxon matched paired test was used for nonparametric analysis of paired data; *p < 0.05, **p < 0.01 as compared with basal CGRP release (n = 6–8).
The effect of levcromakalim and diazoxide on the in vitro release of CGRP from rat TNC. (A) levcromakalim (1 µM) and (B) diazoxide (10 µM) failed to release CGRP above basal levels. (C) Levcromakalim (LK) (1 µM) and (D) diazoxide (10 µM) (D) failed to inhibit CGRP release induced by capsaicin (0.1 µM) (Cap). Values are given as mean ± SEM. The Wilcoxon matched paired test was used for nonparametric analysis of paired data; *p < 0.05, **p < 0.01 as compared with basal CGRP release (n = 5–8).
The final concentrations of ethanol (0.01%) and DMSO (0.01% and 0.1%) in the capsaicin, levcromakalim and diazoxide solutions, respectively, has no effect on basal CGRP release (Saurabh Gupta, personal communication).
Mast cell degranulation studies
Levcromakalim and diazoxide (10−5 M) failed to initiate the degranulation of rat dural mast cells in situ (Figure 6A). Likewise, both drugs (10−8 to 10−4 M) failed to degranulate rat peritoneal mast cells in vitro (Fig 6B). The mast cell degranulator Compound 48/80 (10−5M and 10−4 M, in dura mater and peritoneal mast cells, respectively) was used as a control drug to verify that mast cell degranulation was possible in the two assays.
(A) Degranulation of rat dural mast cells in the presence of levcromakalim (10−5 M) and diazoxide (10−5 M). (B) Degranulation of peritoneal mast cells in the presence of levcromakalim (10−8 to 10−4 M) and diazoxide (10−8 to 10−4 M). The mast cell degranulator Compound 48/80 (10−4M) was used as a control drug to verify both assays. Values are given as mean ± SEM, n = 6–7.
Discussion and conclusions
The main result of the present study is that most subunits of the ATP-sensitive potassium (KATP) channel are present in the trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC), and that KATP channel openers do not induce headache via CGRP release and mast cell degranulation.
mRNA and protein expression studies
We identified the mRNA transcripts and proteins of KATP channel subunits SUR1, Kir6.2, Kir6.1 and SUR2B in the rat TG and TNC, while SUR2A, predominantly found in the sarcolemma of cardiac myocytes, was absent (Figures 1 and 2). In both TG and TNC, SUR2B was weakly detected at the protein level and this is probably explained by the fact that this subunit is solely of vascular origin. While not systematically studied in TG and TNC, two tissues involved in migraine pathophysiology, the distribution of SUR1 and Kir6.2 mRNA has been demonstrated in the mouse brain (11), and Kir6.1 and Kir6.2 immunoreactivity has been shown in structures of the rat brain (12). In central neurons and astrocytes, the subtype of KATP channels expressed is controversial. Some studies report that Kir6.1/SUR1 forms the astrocyte KATP channels (12–14), but Kir6.2 has also been identified there (15). In brain neurons Kir6.2/SUR1 appears to be the major subtype expressed (12–14,16), but Kir6.1 has also been identified (14). Our studies showed expression of both Kir6.1 and Kir6.2 in the rat TG and TNC. When inspecting the western blots, multiple SUR1 protein bands were detected in rat TG and TNC samples (Figure 2A). We suggest these bands identify differentially glycosylated mature SUR1 proteins. SUR1 glycosylation is essential for the proper trafficking and plasma membrane expression of functional SUR1 subunits, and SUR1 glycosylation mutants are retained in the endoplasmic reticulum (17). We were also able to enzymatically deglycosylate SUR1 proteins of different sizes from several rodent tissues (Figure 2B). Thus the appearance of several protein bands on the western blots from both TG and TNC protein lysates suggests that these tissues express fully functional SUR1.
CGRP release
The neuropeptide CGRP is a key molecule in migraine because of its ability to provoke migraine attacks in migraineurs, and its antagonists are effective in migraine treatment (18,19). We found that the SUR2-selective KATP channel opener levcromakalim had no effect on basal CGRP release from dura mater, TG and TNC in vitro (Figures 3, 5). Furthermore, the SUR1-selective KATP channel opener diazoxide also failed to release CGRP from TG and TNC (Dipak Amrutkar, personal communication). Previously it has been shown that activation of KATP channels in neurons induce membrane hyperpolarization and reduces excitability (9,20), and activation of presynaptic KATP channels directly decreases neurotransmitter release from nerve terminals (21–23). Specifically, it has been shown that cromakalim, the racemate of levcromakalim, and pinacidil reduce γ-aminobutyric acid (GABA) release (24). In our studies the KATP channel openers neither potentiated nor inhibited capsaicin-evoked release of CGRP. Furthermore, in a pilot study the KATP channel blocker glibenclamide had no effect on basal CGRP release. Thus, opening of KATP channels has no influence on CGRP release in these migraine related structures.
Mast cell degranulation
The mast cell degranulation assay also failed to show any degranulation effect of levcromakalim and diazoxide (Figure 6). Dural mast cell degranulation has been associated with sensitization of meningeal nociceptors and activation of the trigeminal pain pathway underlying headaches such as migraine (25,26). We therefore explored whether or not KATP channel openers would act directly on mast cells to initiate degranulation and thereby potentially headache. In parallel we also explored whether levcromakalim and diazoxide would degranulate rat peritoneal mast cells, because this assay, as opposed to the dural mast cell assay, is a high-throughput and convenient assay that allowed us to perform concentration-response studies. Moreover, when exposed to an array of different drugs, the outcomes of rat dural and peritoneal mast cell degranulation assays are identical in our hands. However, our studies showed that neither CGRP release nor dural mast cell degranulation are involved in the events used by KATP channel openers to provoke headache.
KATP channels in migraine and pain modulation
Synthetic KATP channel openers such as levcromakalim and pinacidil induce headache as an adverse side effect in clinical studies suggesting that KATP channel opener mechanisms are involved in headache pathology and potentially migraine pain. However, accumulating evidence shows that opening of neuronal KATP channels is an endpoint event in antinociceptive mechanisms in animal models of nociception. Intracerebroventricular administration of cromakalim produces dose-dependent supraspinal antinociception (27,28), and central administration of cromakalim and pinacidil has been shown to potentiate the antinociception induced by subcutaneous morphine (28–30). In addition, it has been demonstrated that powerful antinociceptive agents, including morphine and methadone, reduce neuronal excitability and antinociception through activation of KATP channels at supraspinal, spinal and peripheral levels (28,31). Evidence even suggests that KATP channel openers may mediate their antinociceptive effects via central release of endogenous opioids (32,33). Thus, synthetic KATP channel openers infused during clinical trials provoke headache by a mechanism that does not seem to involve the direct interaction with sensory neuronal KATP channels. Therefore they may produce antinociception only when administered centrally, not peripherally. Among the neuronal structures of the trigeminal system, the TG is located outside the blood-brain barrier, and it is therefore freely accessible to the classic KATP channel openers, because studies suggest that they do not cross the blood-brain barrier under physiologic conditions (33,34). This is in accordance with our observations in rat. We have previously shown that levcromakalim and the potent KATP channel opener P-1075 dilate rat middle cerebral arteries and basilar arteries when applied abluminally, but not luminally, to arteries mounted in a pressure myograph (7). Moreover, KATP channel openers failed to dilate cerebral arteries in vivo when administered through the femoral vein (8), suggesting that these drugs do not readily cross the blood brain barrier in rat. Thus, during clinical trials, KATP channel openers may only directly affect dural and TG structures of the trigeminal pain pathway.
In summary, we suggest that KATP channel openers such as levcromakalim and pinacidil do not induce headache pain through direct interaction with primary sensory neuronal KATP channels in the trigeminal pain pathway. Instead, a vascular-type headache may be initiated through interaction with the Kir6.1/SUR2B KATP channel subtype in the extracerebral tissues.
Footnotes
Funding
This study was supported by grants from the Lundbeck Foundation via Center for Neurovascular Signaling (LUCENS), the Danish Research Council (271-06-0519), the Region of Copenhagen, the NeuroCluster (Faculty of Health Sciences, University of Copenhagen), Torben and Alice Frimodts Foundation, Købmand Sven Hansen and hustru Ina Hansens Foundation, The Illum Foundation and the Ulla and Mogens Folmer Andersens Foundation.
Conflicts of interest
None.